Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Farid Khallouki and Version 2 by Lindsay Dong.
Breast cancer (BC) is the most common female cancer in terms of incidence and mortality worldwide. Tamoxifen (Nolvadex) is a widely prescribed, oral anti-estrogen drug for the hormonal treatment of estrogen-receptor-positive BC, which represents 70% of all BC subtypes.
- tamoxifen
- estrogen receptor
- breast cancer
1. Tamoxifen and Its Interaction with the ER
Estrogens, primarily produced in the ovarian follicles and adrenal glands, are the most important female sex hormones [1][15]. The estrogen receptor (ER) is a member of the nuclear receptor superfamily; its function, which is mediated by 17β-estradiol (E2), is to activate the transcription of genes involved in the growth and differentiation of cells, as well as reproduction.
The two distinct subtypes of ER, ERα and ERβ, each have different effects in gene regulation and cell proliferation. ERα is mainly present in tissues with reproductive functions, such as the ovaries, uterus, and breast, along with the brain, heart, liver, and bone. ERβ is detected in the ovarian, uterine, breast, hypothalamic, and cardiovascular tissues [2][16].
Structurally, ERs consist of six functional domains: A, B, C, D, E, and F, with three major domains including an N-terminal (A/B domain), a DNA-binding domain (C domain), and a ligand-binding domain (LBD, E domain) (Figure 1). The N-terminal (A/B domain) activation function (AF-1) region is involved in protein–protein interactions, is hormone-independent, and is mostly regulated by phosphorylation. The C-terminal (E domain) activation function (AF-2) contains the binding region for the ligand, along with co-activators or co-repressors, and is responsible for ligand-gated transcription. The highly conserved DNA-binding C domain consists of two zinc finger modules, which establishes contacts with specific response elements and is involved in the spatial recognition of the receptor. The flexible hinge or D domain contains a nuclear localization signal, acting as a dimerization region. Lastly, the F domain is variable, and its function is still poorly understood [1][15].
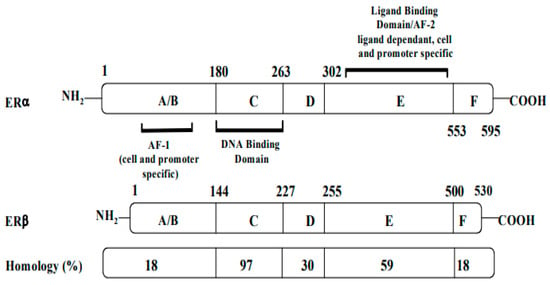
Figure 1.
Structures, functional regions, and percentage of sequence homology of two human estrogen receptors: ERα and ERβ.
Both receptors are organized in six functional domains: the A/B domain at the N-terminal, the C domain of the DNA-binding domain (DBD), the D domain of the nuclear translocation signal, and the E/F domain at the C-terminus including the ligand-binding domain (LBD) and the ligand-dependent activation function AF-2.
Although the two ERs possess different functions, they share a homologous DNA-binding domain and a 55–60% homology in their ligand-binding domains (LBDs). Both coexist in breast tissues, with more ERα than ERβ in breast tumors [3][17]. While AF-1 has only 20% homology in ERα and ERβ, AF-2 is especially similar in both ERs [4][18].
In the presence of natural or synthetic agonists of the ER, AF-1 and AF-2 domains will interact with steroid receptor coactivator proteins. These include the p160/steroid receptor coactivator (SRC) family, which comprises three pleiotropic coregulators (SRC-1, SRC-2, and SRC-3) that serve as the transcription factors that regulate the expression of genes [5][6][19,20]. Hormone-activated receptors form multiple dimers of αα monomers, ββ monomers, and a combination of αβ monomers [7][21]. Depending on the composition of the dimers, cell homeostasis regulation and the affinity for different estrogen response elements (EREs) might vary.
The selective action of the ER might depend on the structure of the ligand, the type or isotype of ER, the type of co-regulator, and the interaction of the complex with EREs. Figure 2 depicts the action of ER in terms of explaining the tissue-specific binding of E2 to its receptors.
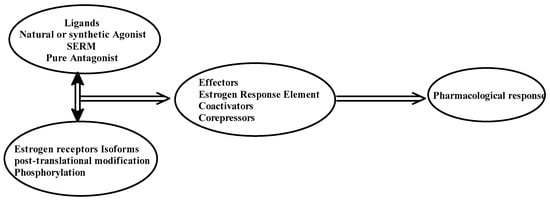
Figure 2.
Pharmacological action of estrogen receptors.
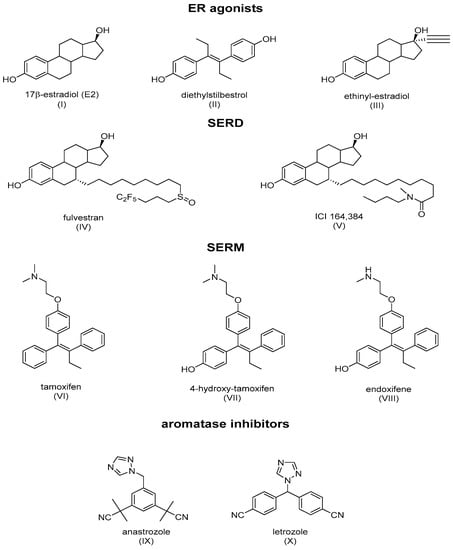
ER ligands are classified into four families: ER agonists, SERDs, SERMs, and aromatase inhibitors (Figure 3). ER agonists include endogenous natural 17β-estradiol (Figure 3I), diethylstilbestrol (Figure 3II), and ethinylestradiol (Figure 3III). In addition, 17β-estradiol (E2) agonizes ERs in all tissue types where they are expressed and recruit coactivators to activate gene expression under the control of EREs, thereby promoting DNA synthesis and the proliferation of ER-responsive cells. Selective degraders of estrogen receptors (SERDs) completely inhibit and degrade ERα. Examples include the synthetic drugs ICI 182,780 (Fulvestrant) (Figure 3IV) and ICI 164,384 (Figure 3V), with the systematic name of N-n-butyl-N-methyl-11-[3,17β-dihydroxy-estra-1,3,5(10)-trien,7α-yl]-undecanamide. Selective modulators of estrogen receptors (SERMs) are unique in their anti-estrogen activity, depending on the type of tissue. Mounting evidence supports the unique characteristics of SERM activity, which is primarily determined by the selective recruitment of both ERα repressors and activators into specific tissues [4][8][18,22]. The main example is tamoxifen (TAM), which is a partial agonist of the first generation of SERMs (Figure 3VI). The final class of drugs, including anastrozole (Figure 3IX) and letrozole (Figure 3X), are aromatase inhibitors (AIs). AIs inhibit the aromatization of androgens at the CYP9A1 level, and block the endogenous formation of estrogens.
Figure 3. Main classes of agents used in hormone therapy that target estrogen signaling: ER agonists, SERDs (Fulvestran and ICI 164,384), SERMs (tamoxifen), and aromatase inhibitors.
TAM acts by blocking the mitogenic action of E2, via competition with E2 on ERα [9][10][26,27]. The structural analyses of ERα-4OHTam were conducted and showed that the drug effectively accommodates the ER ligand-binding pocket [11][12][13][28,29,30]. OH-TAM induced conformational changes that were different from E2 on ER-LBD, which affected the recruitment of the co-regulators and the regulation of gene expression [14][31]. The LBD, which consists of amino acids 304 to 553 of the ER, is composed of a cluster of twelve α-helices (H1–H12), which harbor a highly structured ligand-dependent domain containing dimerization interfaces, co-activation and co-repression functions, and a hydrophobic pocket that accommodates hormones.
ERα, in the absence of hormones, is inactivated as a complex by several chaperone proteins, such as HSP90, HSP70, and HSP40, with the aid of several other co-chaperones including cyclophilin-40 and p23 proteins. These heat shock proteins help to maintain the receptor in an adequate conformation, allowing for it to respond quickly to a hormonal signal. The binding of the hormone to the receptor results in the release of chaperonins and induces the nuclearization of the ER-E2 complex, which is followed by dimerization and stabilization in a conformation where the last helix (H12) folds over the ligand-binding pocket (LBP) and forms a hydrophobic groove, sealing the LBP [15][32].
In the presence of agonists, helix 12 traps the ligand inside the hydrophobic pocket of the ligand-binding domain. Helix 12’s hydrophobic grooves become exposed to the nuclear box recognition sequence containing the key LXXLL motif, which is rich in leucine. Alternatively, SERM drives the hydrophobic pocket into an open state, which dislocates helix 12 from forming an active AF-2 conformation, and specifically occupies the space for a coregulatory protein recognition sequence of LXXLL binding, thereby blocking the coactivators from binding and preventing gene transcription. The crystal structures of LBD complexed with 4-hydroxy-tamoxifen and ICI-164, 384 (Figure 3V) (pure antiestrogen) reveal evidence of certain critical parameters with regard to these interactions. Figure 4 depicts the mechanism of action of the ER ligands at the ERα level. E2, TAM, and ICI 164,384 (ICI) bind to ER LBD, which explains why they compete with E2 at the ERα level and can inhibit E2-mediated BC cell proliferation. TAM might exert its antiproliferative potency through other ER-dependent mitogenic pathways [16][17][18][19][20][33,34,35,36,37]. TAM also possesses ERα-independent antiproliferative properties and displays a certain degree of efficacy in the context of ER-negative cancer cells (see vide infra).
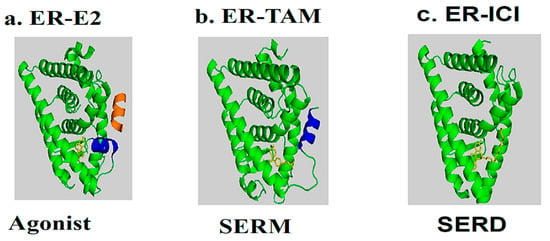
Figure 4. Molecular model and ribbon representation highlighting the action mechanism of estrogen receptors and the positioning of helix H12. (a) ER-E2 represents estradiol (yellow) in the presence of a co-activator (orange). (b) ER-TAM shows ER-LBD with tamoxifen (yellow), which is conducted to induce co-repression (SERM). (c) The ER-ICI interaction shows the full antagonist activity (SERD) in the absence of helix H12. Helix H12 in (a,b) is drawn as a cylinder in blue (Poirot et al., unpublished data).
2. Insight to the Chemo-Preventive Action of Tamoxifen in Breast Cancer
Chemo-prevention is an oncological prophylaxis that modulates the process of carcinogenesis in the early stages of cancer development with the use of pharmacological agents and non-nutritional food components [21][38]. Since the 1970s, TAM has been a landmark form of BC treatment. It was initially recommended as an antiestrogen to help treat hormone-responsive BC, but its curative potential was later expanded to adjuvant therapy in order to help reduce the incidence of BC. TAM inhibits cell proliferation through its actions on the ERα, which regulates the processing of growth factors. This can lead to the arrest of cell growth, or cell death and tumor regression. TAM was, therefore, shown to be an efficient chemo-preventive agent in both postmenopausal and premenopausal women with a moderate risk of BC [22][39]. The results of meta-analyses of randomized clinical trials and case–control studies reported a decrease in BC incidence in women receiving TAM as a postoperative adjuvant compared to untreated women [23][24][25][26][27][28][29][30][31][6,40,41,42,43,44,45,46,47].
Adjuvant TAM is commonly administered for 5 years; however, 10 years of TAM therapy has been associated with a greater efficacy in patients with ER-positive BC. In addition, this longer duration of therapy might reduce mortality caused by BC during the second decade after diagnosis by half [30][32][46,48]. Moreover, TAM reduces the risk of developing ER-positive BC by 48% in women over 35 years old [33][49]. Although the main rationale in the clinical use of TAM is the blockage of mitogenic actions of E2, several other mechanisms of TAM have been observed that could account, in part, to its anticancer action. Furthermore, TAM has been shown to induce cell death, which has been earlier summarized in certain other excellent reviews [34][35][36][50,51,52].
TAM binds with a nanomolar affinity to a microsomal binding site, called the anti-estrogen binding site (AEBS) [37][53]. Selective AEBS ligands include the diphenyl methane compounds PBPE (Figure 5I) and DPPE (tesmilifene, Figure 5II). AEBS ligands include SERMs, bearing a cationic amino alkyl side chain; estrogens and pure antiestrogens, on the other hand, have no affinity for the AEBS. Oxysterols (e.g., 6-ketocholestanol (Figure 5III), 7-ketocholestanol (Figure 5VI), and 7-ketocholesterol (Figure 5V), as well as unsaturated fatty acids and histamine), have also been characterized as AEBS ligands [35][51]. AEBS is found in various tissues, including BC cells, and is independent of ER status. Kedjouar et al. reported that AEBS is a multiproteic complex composed of two cholesterogenic enzymes: 3β-hydroxysterol-Δ8-Δ7-isomerase (EBP/D8D7I) and 3β-hydroxysterol-Δ7-reductase (DHCR7) [38][54]. It was later shown that the AEBS carries out cholesterol-5,6-epoxide hydrolase (ChEH) enzymatic activity that catalyzes the hydrolysis of 5,6-epoxycholestan-3β-ol diastereomers (5,6α-EC (Figure 5VI) and 5,6β-EC (Figure 5VII) into cholestane-3β,5α,6β-triol (CT) (Figure 5VIII) [39][40][55,56].
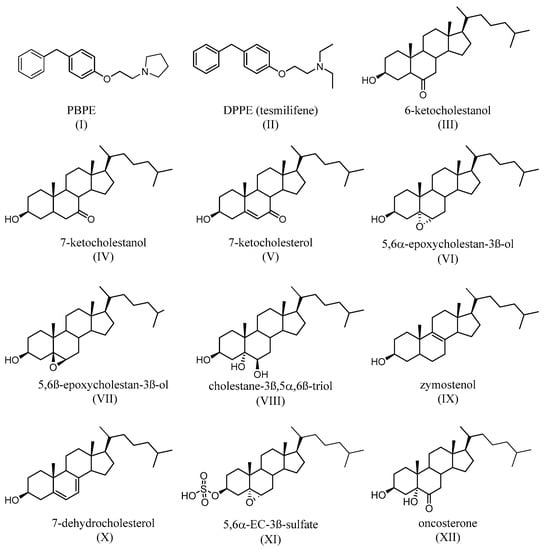
Figure 5. Synthetic (I,II) and endogenous (III–V) selective AEBS ligands with no affinity for ER. Oxysterols that accumulate after the inhibition of ChEH by AEBS ligands (VI–VII). Product of the ChEH enzymatic activity (VIII). Cholesterol precursors that can accumulate after the binding of ligands (including SERMs) on the AEBS (IX,X). The signaling sterol that accumulates in cells expresses SULT2B1b, after ChEH inhibition by AEBS ligands (XI). The tumor promoter whose biosynthesis is blocked by AEBS ligands (including SERMs) (XII).
TAM, SERMs, and the selective AEBS ligand PBPE (Figure 5I) induce the intracellular accumulation of cholesterol precursors, including zymostenol (Figure 5IX), the substrate of D8D7I, and 7-dehydrocholesterol (Figure 5X), which is the substrate of DHCR7. AEBS ligands also increase the total sterol levels in the cell [41][57]. Interestingly, accumulated cholesterol precursors undergo oxidation toward unidentified oxysterols. Since AEBS also carries out ChEH activity, the intracellular accumulation of 5,6-EC and the diminution of CT (Figure 5VIII) have thus been described, on BC cells, as being exposed to AEBS ligands. In addition, CT is metabolized by 11β-hydroxysteroid-dehydrogenase type 2 into oncosterone (Figure 5XII), an oncometabolite that promotes BC development [42][58]. Consequently, targeting ChEH with AEBS ligands inhibits both the production of CT and oncosterone in BC cells. It was shown that the inhibition of oncosterone production represents a promising approach for the development of new anticancer agents [42][43][44][58,59,60].