Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jiemei Wang | -- | 2050 | 2023-06-06 17:48:05 | | | |
| 2 | Conner Chen | Meta information modification | 2050 | 2023-06-07 07:27:36 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Minjares, M.; Wu, W.; Wang, J. Generation of Reactive Oxygen Species in Endothelial Cells. Encyclopedia. Available online: https://encyclopedia.pub/entry/45255 (accessed on 10 August 2026).
Minjares M, Wu W, Wang J. Generation of Reactive Oxygen Species in Endothelial Cells. Encyclopedia. Available at: https://encyclopedia.pub/entry/45255. Accessed August 10, 2026.
Minjares, Morgan, Wendy Wu, Jie-Mei Wang. "Generation of Reactive Oxygen Species in Endothelial Cells" Encyclopedia, https://encyclopedia.pub/entry/45255 (accessed August 10, 2026).
Minjares, M., Wu, W., & Wang, J. (2023, June 06). Generation of Reactive Oxygen Species in Endothelial Cells. In Encyclopedia. https://encyclopedia.pub/entry/45255
Minjares, Morgan, et al. "Generation of Reactive Oxygen Species in Endothelial Cells." Encyclopedia. Web. 06 June, 2023.
Copy Citation
Reactive oxygen species (ROS) are radical oxygen intermediates that serve as important second messengers in signal transduction. However, when the accumulation of these molecules exceeds the buffering capacity of antioxidant enzymes, oxidative stress and endothelial cell (EC) dysfunction occur.
endothelial cells
oxidative stress
ROS
1. Introduction
Reactive oxygen species (ROS) are byproducts of enzymatic reactions, composed of both free radical and non-free radical oxygen intermediates, and are generated in numerous cell compartments such as the endoplasmic reticulum (ER), mitochondria, and cell membrane [1][2]. At physiological concentrations, ROS are generated by several sources, including uncoupled endothelial nitric oxide synthases (eNOS), NADPH oxidases (NOX), xanthine oxidases (XO), cyclooxygenases, and the mitochondrial electron transport chain [3]. Antioxidant enzymes such as superoxide dismutase (SOD), catalase (CAT), and peroxidase (POD) are required to manage the production of ROS for cellular health [4][5].
Cardiovascular (CV) diseases, the leading causes of death worldwide, act to remodel blood vessels and restrict blood flow to the heart and nervous system [6]. Oxidative stress contributes to the development of CV risks and disorders, including hypertension, atherosclerosis, diabetes mellitus (DM), cardiomyopathy, obesity, and congestive heart failure [7][8]. The endothelium not only regulates the passage of nutrients between tissues, forms the inner lining of all blood vessels, and manages the protective barrier properties of the vascular system [9][10], but also modulates vascular growth and permeability, tissue metabolism, immune responses to inflammatory stimulation, stem cell recruitment, and regulation of vascular tone [11][12]. Endothelial cells (ECs) also secrete a wide range of cytokines and growth factors that regulate various physiological activities in autocrine, paracrine, and endocrine manners. One of the important molecules that ECs produce is nitric oxide (NO), generated by endothelial NO synthase (eNOS). NO is a vasoactive molecule that participates in vascular remodeling, vasodilation, platelet aggregation and adhesion, clot formation, and renal hemodynamics [13]. ROS serve as important second messengers for signal transduction and aid in modulating EC activation, proliferation, and angiogenesis [14][15]. However, under CV risks or disorders when the accumulation of ROS exceeds the buffering capacity of antioxidant enzymes, oxidative stress and molecular damage occur, leading to early apoptotic death and genomic alteration in ECs [16]. ECs then switch from a vasodilative, anti-inflammatory, anticoagulant environment to a vasoconstrictive, proinflammatory, and procoagulant environment, with decreased bioavailability of nitric oxide (NO) and overproduction of superoxide (O2−), leading to impaired cellular repair [17][18] and endothelial dysfunction.
Extensive studies have investigated the therapeutic potential of various antioxidant therapies in ECs [6], with recent studies turning to the investigation of microRNAs (miRNA). MicroRNAs are short, non-coding RNA molecules that silence post-transcriptional target genes by promoting messenger RNA (mRNA) cleavage and/or inhibition of protein translation [19][20]. Emerging evidence has suggested that ROS and microRNAs co-regulate each other. ROS dysregulate the expression of miRNAs in vascular and immune cells, while miRNA manages oxidative stress by targeting specific mRNAs [21]. Due to the biodistribution of these molecules and the presence of multiple mRNA targets for a single microRNA, mechanistic approaches for reducing excessive oxidative stress without impacting other pathways have been evolving [22].
2. Generation of ROS in Endothelial Cells
ECs actively respond to hemodynamic changes and blood-borne signals and facilitate the exchange of oxygen, nutrients, solutes, hormones, and macromolecules between the blood and surrounding tissues, making them essential in the management of metabolic homeostasis [23]. Production of ROS by ECs is a required process for normal cellular function and can be triggered by oxidants produced by activated immune cells, cytokines, and other physical stimuli [23]. Intracellular ROS consist of superoxide, hydrogen peroxide (H2O2), and hydroxyl radicals that are derived from uncoupled eNOS, NADPH oxidase, electron transport chain in mitochondria, and xanthine oxidase (Figure 1) [24][25]. Under controlled environments, these ROS serve as second messengers regulating cell proliferation, differentiation, immune responses, and tissue repair [26]. However, an overabundance of ROS leads to EC dysfunction, increasing the risk of atherosclerosis, hypertension, hypercholesterolemia, stroke, diabetes, obesity, and other metabolic syndromes [27][28].
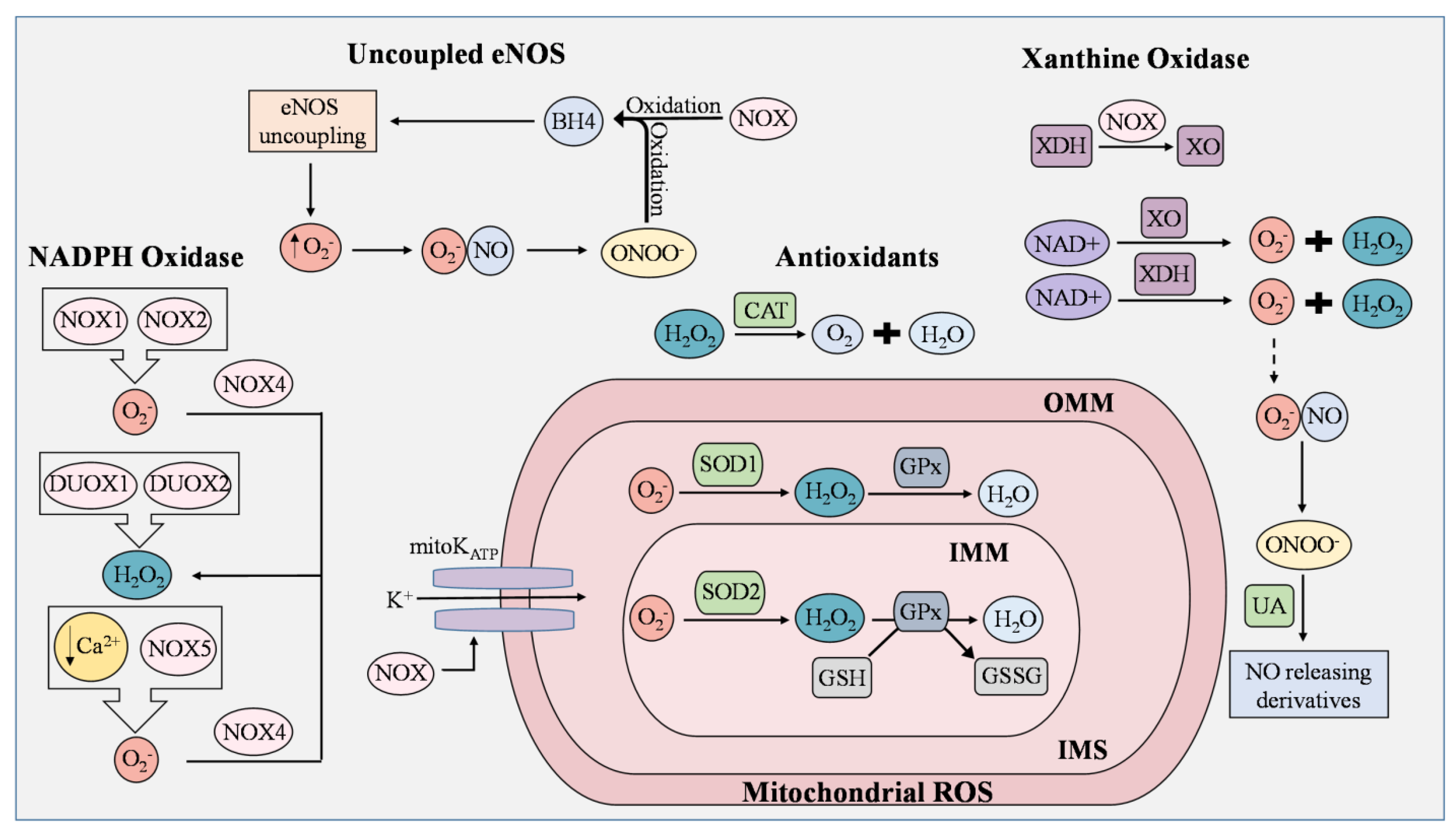
Figure 1. ROS generation and removal in endothelial cells. Reactive oxygen species (ROS) are mainly derived from uncoupled endothelial nitric oxide synthase (eNOS), NADPH oxidase (NOX), mitochondria, and xanthine oxidase (XO). Antioxidants that regulate ROS include catalase (CAT), superoxide dismutase 1 (SOD1), superoxide dismutase 2 (SOD2), and uric acid (UA). During eNOS uncoupling, superoxide (O2−) is produced in abundance rather than nitric oxide (NO). Superoxide then reacts with NO to generate peroxynitrite (ONOO−), which rapidly oxidizes tetrahydrobiopterin (BH4), thereby maintaining the state of eNOS uncoupling. ROS produced from NOX isoforms are involved in proliferation, migration, and differentiation in ECs, and are known to cause mitochondrial DNA damage, induce oxidative inactivation of BH4, stimulate the conversion of xanthine dehydrogenase (XDH) to XO, and manipulate the opening of the mitochondrial ATP-sensitive K+ ion channel (mitoKATP). NOX1 and NOX2 generate superoxide as opposed to NOX4, which converts superoxide to H2O2. In low concentrations of Ca2+, NOX5 enhances ROS production in ECs. Dual oxidase 1 (DUOX1) and dual oxidase 2 (DUOX2) possess roles in immune response and cell differentiation and are responsible for H2O2 generation. Leakage of electrons from the mitochondrial electron transport chain generates superoxide, but the presence of SOD1 and SOD2 prevents the buildup of damaging mitochondrial ROS (mROS). In the inner mitochondrial matrix (IMM), superoxide is converted to H2O2 by SOD2, while superoxide in the intermembrane space (IMS), which is between the IMM and outer mitochondrial matrix (OMM), is converted by SOD1. H2O2 is reduced to H2O by oxidizing glutathione (GSH) into glutathione disulfide (GSSG). This reaction is catalyzed by glutathione peroxidase (GPx). Catalase is an Fe-containing enzyme that catalyzes hydrogen peroxide into H2O and molecular oxygen. XO and XDH are the two interconvertible forms of xanthine oxidoreductase (XOR) where NAD+ is used to generate superoxide and peroxide. Under normal physiological conditions, UA possesses antioxidant activity and can aid in the regulation of ROS generated by XOR by reacting with ONOO− to form NO-releasing derivatives.
2.1. Uncoupled eNOS
In ECs, the eNOS enzyme produces NO, an unorthodox, highly reactive messenger molecule that regulates vascular tone, gene transcription, mRNA translation, and post-translational modifications of proteins [29]. eNOS requires L-arginine and molecular oxygen (O2) to produce NO along with the cofactors tetrahydrobiopterin (BH4), nicotinamide adenine diphosphate (NADPH), flavin mononucleotide, heme, and flavin adenine dinucleotide [30]. Depletion of these molecules leads to eNOS uncoupling, where O2− is produced in abundance rather than NO. Superoxide reacts with NO to generate peroxynitrite (ONOO−). This unstable molecule rapidly oxidizes BH4, which then maintains the state of eNOS uncoupling and reduces NO bioavailability [30][31]. This rapid oxidative inactivation of NO and overproduction of ROS by uncoupled eNOS has been shown to increase the risk of developing vascular disease factors [32][33].
2.2. NADPH
NADPH oxidase is an enzyme whose primary function is to catalyze the production of superoxide from oxygen and NADPH. ROS produced from NOX isoforms are involved in proliferation, migration, and differentiation in ECs [34][35], and are known to cause mitochondrial DNA damage, induce oxidative inactivation of BH4, stimulate the conversion of xanthine dehydrogenase (XDH) to XO, and manipulate the opening of the mitochondrial ATP-sensitive K+ ion channel (mitoKATP) [35]. The NOX family has seven isoforms, known as NOX1/2/3/4/5 and dual oxidase 1/2 (DUOX1/2). Many have been found to induce vascular dysfunction and inflammation [36]. NOX1 and NOX2 generate superoxide as opposed to NOX4, which is constitutively active and converts superoxide to H2O2 (Figure 1) [37][38]. In addition, NOX2 oversees the transfer of electrons from cytosolic NADPH to molecular oxygen and participates in signal transduction, angiogenesis, and cell death [39]. NOX3, although also responsible for generating ROS, is mainly expressed in the inner ear and has not been observed as a key factor in EC dysfunction and vascular damage [40]. NOX5 is the only calcium-regulated isoform present in the endothelium and has been found to interact with glucose and angiotensin II [41]. In low concentrations of Ca2+, NOX5 enhances ROS production in ECs [39]. DUOX1 and DUOX2 possess roles in immune response and cell differentiation, with both being expressed in the stomach, lungs, and thyroid. Their functions in the vascular system are relatively uncertain, although they are responsible for H2O2 generation [42]. Overall, these enzymes serve as primary sources of ROS in ECs and require further investigation in metabolic disorders and the vascular system.
2.3. Mitochondrial Electron Transport Chain
Mitochondria are crucial for the generation of ATP in cells. In ECs, mitochondrial-derived reactive oxygen species (mROS) are critical for cellular responses to vascular risk factors. Leakage of electrons from the mitochondrial electron transport chain generates superoxide. The production of mROS by the ETC is tightly regulated in order to avoid oxidative damage to cellular processes [43]. In the inner membrane, electron transport chain complexes I-IV generate a proton gradient that drives the production of ATP [25]. First, electrons (e−) from NADH and FADH2 pass through complexes I and II into complex III by ubiquinol (CoQ). Cytochrome c then transfers the electrons to complex IV, where O2 is reduced into H2O. Protons reenter the mitochondrial matrix through complex V and are used to generate ATP [44]. During this cycle, electrons are leaked from complexes I and III, and superoxide is then generated toward the matrix and intermembrane space [44][45]. The increase in superoxide results in the swelling of mitochondria and further membrane breakdown, leading to the release of cytochrome C. The latter then triggers a caspase cascade and induces cell apoptosis [46]. The mitochondria dysfunction of this organelle is one of the major causes of ROS overproduction and EC dysfunction, making it a potential target for CVD treatments.
2.4. Xanthine Oxidases
Xanthine oxidoreductase (XOR) is a terminal enzyme that produces superoxide and hydrogen peroxide by catalyzing the oxidation of hypoxanthine, which is then converted to uric acid (UA) [47][48]. XO and xanthine dehydrogenase (XDH) are the two interconvertible forms of XOR where NAD+ is used to generate superoxide and peroxide [49][50]. Though this enzyme is one of the major sources of ROS, it is mainly studied due to its ability to produce uric acid. UA is the end-product of purine metabolism. Under normal physiological conditions, UA possesses antioxidant activity and can aid in the regulation of ROS generated by XOR by reacting with ONOO− to form NO-releasing derivatives or by targeting the Nrf2 pathway [51][52][53]. When synthesis and excretion are unbalanced, hyperuricemia can occur [51] and cause inflammation and EC dysfunction, eventually progressing into gout, atherosclerosis, or chronic kidney disease if left untreated [53]. Although an increase in XOR levels can lead to damaging ROS, the conversion of these molecules into uric acid can provide oxidative benefits.
2.5. Antioxidant/Defense Systems in ECs for ROS
Antioxidants counteract and eliminate ROS, making them critical molecules to defend against oxidative stress in ECs. The two types of antioxidants include enzymatic and non-enzymatic, where enzymatic antioxidants directly catalyze ROS and non-enzymatic either promote anti-oxidative enzymes or aid in oxidative chain reactions [54]. Superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx) are the main enzymes involved in ROS metabolism. SODs, which catalyze the conversion of superoxide to hydrogen peroxide, are composed of three isoforms, including copper–inc superoxide dismutase (Cu/ZnSOD, SOD1), manganese superoxide dismutase (MnSOD, SOD2), and extracellular superoxide dismutase (EcSOD, SOD3) [55]. Cu/ZnSOD is an active dimer that converts superoxide into molecular oxygen and hydrogen peroxide using copper and zinc as its metal cofactors [56]. MnSOD, the greatest source of cellular ROS of the three isoforms, regulates ROS in the mitochondrial matrix [57]. EcSOD is the least-studied isoform but has thus far been shown to exert anti-inflammatory abilities, regulate cell barrier function, modulate cytokine response, and protect tissues against ROS [58]. In the mitochondrial matrix, superoxide is converted to H2O2 by SOD2, while intermembrane space superoxide is converted by SOD1 after it passes through voltage-dependent anion channels (VDAC) [59]. Catalase is an Fe-containing enzyme that catalyzes hydrogen peroxide into H2O and molecular oxygen [60]. Heme-containing enzymes, true catalases, catalase-peroxides, and manganese catalases are used to classify this enzyme [61]. Yang et al. found that inhibiting catalase in ECs promotes ROS production and enhances oxidative damage under a hypoxic environment [62]. Similar to catalase, GPx is also responsible for reducing hydrogen peroxide and catalyzing GSSG, with GSH as a reductant [63][64]. Prasai et al. inhibited glutathione reductase activity and observed enhanced protein S-glutathionylation, VEGFR2 activation, and increased reactive oxygen species (ROS) production in aortic ECs, demonstrating that the cellular GSH: GSSG ratio is critical in managing oxidative stress [65]. Antioxidant enzymes can be impaired in many metabolic disorders, particularly in hyperglycemia, due to multifactorial mechanisms [66]. They have become therapeutic targets as restoration of their ability to reduce oxidative stress and prevent EC dysfunction provides a path toward reducing the risk of developing cardiometabolic disorders.
References
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057–1073.
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902.
- Villalpando-Rodriguez, G.E.; Gibson, S.B. Reactive Oxygen Species (ROS) Regulates Different Types of Cell Death by Acting as a Rheostat. Oxid. Med. Cell. Longev. 2021, 2021, 9912436.
- Celotto, A.M.; Liu, Z.; Vandemark, A.P.; Palladino, M.J. A novel Drosophila SOD2 mutant demonstrates a role for mitochondrial ROS in neurodevelopment and disease. Brain Behav. 2012, 2, 424–434.
- Liu, N.; Lin, Z.; Guan, L.; Gaughan, G.; Lin, G. Antioxidant enzymes regulate reactive oxygen species during pod elongation in Pisum sativum and Brassica chinensis. PLoS ONE 2014, 9, e87588.
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864.
- Taverne, Y.J.; Bogers, A.J.; Duncker, D.J.; Merkus, D. Reactive oxygen species and the cardiovascular system. Oxid. Med. Cell. Longev. 2013, 2013, 862423.
- Rotariu, D.; Babes, E.E.; Tit, D.M.; Moisi, M.; Bustea, C.; Stoicescu, M.; Radu, A.F.; Vesa, C.M.; Behl, T.; Bungau, A.F.; et al. Oxidative stress-Complex pathological issues concerning the hallmark of cardiovascular and metabolic disorders. Biomed. Pharmacother. 2022, 152, 113238.
- Bautch, V.L.; Caron, K.M. Blood and lymphatic vessel formation. Cold Spring Harb. Perspect. Biol. 2015, 7, a008268.
- Aman, J.; Weijers, E.M.; van Nieuw Amerongen, G.P.; Malik, A.B.; van Hinsbergh, V.W. Using cultured endothelial cells to study endothelial barrier dysfunction: Challenges and opportunities. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L453–L466.
- Hanson, M.; Gluckman, P. Endothelial dysfunction and cardiovascular disease: The role of predictive adaptive responses. Heart 2005, 91, 864–866.
- Sturtzel, C. Endothelial Cells. Adv. Exp. Med. Biol. 2017, 1003, 71–91.
- Cyr, A.R.; Huckaby, L.V.; Shiva, S.S.; Zuckerbraun, B.S. Nitric Oxide and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 307–321.
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 2018, 100, 1–19.
- Alhayaza, R.; Haque, E.; Karbasiafshar, C.; Sellke, F.W.; Abid, M.R. The Relationship Between Reactive Oxygen Species and Endothelial Cell Metabolism. Front. Chem. 2020, 8, 592688.
- Sinenko, S.A.; Starkova, T.Y.; Kuzmin, A.A.; Tomilin, A.N. Physiological Signaling Functions of Reactive Oxygen Species in Stem Cells: From Flies to Man. Front. Cell Dev. Biol. 2021, 9, 714370.
- Wang, M.; Li, Y.; Li, S.; Lv, J. Endothelial Dysfunction and Diabetic Cardiomyopathy. Front. Endocrinol. 2022, 13, 851941.
- Gallo, G.; Volpe, M.; Savoia, C. Endothelial Dysfunction in Hypertension: Current Concepts and Clinical Implications. Front. Med. 2021, 8, 798958.
- Ho, P.T.B.; Clark, I.M.; Le, L.T.T. MicroRNA-Based Diagnosis and Therapy. Int. J. Mol. Sci. 2022, 23, 7167.
- Lu, T.X.; Rothenberg, M.E. MicroRNA. J. Allergy Clin. Immunol. 2018, 141, 1202–1207.
- K, R.B.; Tay, Y. The Yin-Yang Regulation of Reactive Oxygen Species and MicroRNAs in Cancer. Int. J. Mol. Sci. 2019, 20, 5335.
- Ilieva, M.; Panella, R.; Uchida, S. MicroRNAs in Cancer and Cardiovascular Disease. Cells 2022, 11, 3551.
- Muller, N.; Warwick, T.; Noack, K.; Malacarne, P.F.; Cooper, A.J.L.; Weissmann, N.; Schroder, K.; Brandes, R.P.; Rezende, F. Reactive Oxygen Species Differentially Modulate the Metabolic and Transcriptomic Response of Endothelial Cells. Antioxidants 2022, 11, 434.
- Kiselyov, K.; Muallem, S. ROS and intracellular ion channels. Cell Calcium 2016, 60, 108–114.
- Zheng, D.; Liu, J.; Piao, H.; Zhu, Z.; Wei, R.; Liu, K. ROS-triggered endothelial cell death mechanisms: Focus on pyroptosis, parthanatos, and ferroptosis. Front. Immunol. 2022, 13, 1039241.
- Loperena, R.; Harrison, D.G. Oxidative Stress and Hypertensive Diseases. Med. Clin. N. Am. 2017, 101, 169–193.
- Sun, H.J.; Wu, Z.Y.; Nie, X.W.; Bian, J.S. Role of Endothelial Dysfunction in Cardiovascular Diseases: The Link between Inflammation and Hydrogen Sulfide. Front. Pharmacol. 2019, 10, 1568.
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636.
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837.
- Alkaitis, M.S.; Crabtree, M.J. Recoupling the cardiac nitric oxide synthases: Tetrahydrobiopterin synthesis and recycling. Curr. Heart Fail. Rep. 2012, 9, 200–210.
- Luczak, A.; Madej, M.; Kasprzyk, A.; Doroszko, A. Role of the eNOS Uncoupling and the Nitric Oxide Metabolic Pathway in the Pathogenesis of Autoimmune Rheumatic Diseases. Oxid. Med. Cell. Longev. 2020, 2020, 1417981.
- Forstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735.
- Wu, Y.; Ding, Y.; Ramprasath, T.; Zou, M.H. Oxidative Stress, GTPCH1, and Endothelial Nitric Oxide Synthase Uncoupling in Hypertension. Antioxid. Redox Signal. 2021, 34, 750–764.
- Babior, B.M. NADPH oxidase. Curr. Opin. Immunol. 2004, 16, 42–47.
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194.
- Deliyanti, D.; Alrashdi, S.F.; Touyz, R.M.; Kennedy, C.R.; Jha, J.C.; Cooper, M.E.; Jandeleit-Dahm, K.A.; Wilkinson-Berka, J.L. Nox (NADPH Oxidase) 1, Nox4, and Nox5 Promote Vascular Permeability and Neovascularization in Retinopathy. Hypertension 2020, 75, 1091–1101.
- Schroder, K. Isoform specific functions of Nox protein-derived reactive oxygen species in the vasculature. Curr. Opin. Pharmacol. 2010, 10, 122–126.
- Fulton, D.J.; Barman, S.A. Clarity on the Isoform-Specific Roles of NADPH Oxidases and NADPH Oxidase-4 in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 579–581.
- Vermot, A.; Petit-Hartlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890.
- Zhao, T.; Wang, Y.; Li, Z.; Xu, X.; Lei, S.; Huang, L.; Xu, L.; Zhang, M.; Yang, L. Associations of noise kurtosis, genetic variations in NOX3 and lifestyle factors with noise-induced hearing loss. Environ. Health 2020, 19, 13.
- Marques, J.; Fernandez-Irigoyen, J.; Ainzua, E.; Martinez-Azcona, M.; Cortes, A.; Roncal, C.; Orbe, J.; Santamaria, E.; Zalba, G. NADPH Oxidase 5 (NOX5) Overexpression Promotes Endothelial Dysfunction via Cell Apoptosis, Migration, and Metabolic Alterations in Human Brain Microvascular Endothelial Cells (hCMEC/D3). Antioxidants 2022, 11, 2147.
- Van der Vliet, A.; Danyal, K.; Heppner, D.E. Dual oxidase: A novel therapeutic target in allergic disease. Br. J. Pharmacol. 2018, 175, 1401–1418.
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485.
- Tang, X.; Luo, Y.X.; Chen, H.Z.; Liu, D.P. Mitochondria, endothelial cell function, and vascular diseases. Front. Physiol. 2014, 5, 175.
- Giorgi, C.; Marchi, S.; Simoes, I.C.M.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jedrak, P.; Pierzynowska, K.; et al. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344.
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural. Regen. Res. 2013, 8, 2003–2014.
- Chen, Q.M. Nrf2 for protection against oxidant generation and mitochondrial damage in cardiac injury. Free Radic. Biol. Med. 2022, 179, 133–143.
- Tang, S.P.; Mao, X.L.; Chen, Y.H.; Yan, L.L.; Ye, L.P.; Li, S.W. Reactive Oxygen Species Induce Fatty Liver and Ischemia-Reperfusion Injury by Promoting Inflammation and Cell Death. Front. Immunol. 2022, 13, 870239.
- Casas, A.I.; Nogales, C.; Mucke, H.A.M.; Petraina, A.; Cuadrado, A.; Rojo, A.I.; Ghezzi, P.; Jaquet, V.; Augsburger, F.; Dufrasne, F.; et al. On the Clinical Pharmacology of Reactive Oxygen Species. Pharmacol. Rev. 2020, 72, 801–828.
- Batty, M.; Bennett, M.R.; Yu, E. The Role of Oxidative Stress in Atherosclerosis. Cells 2022, 11, 3843.
- Yu, W.; Cheng, J.D. Uric Acid and Cardiovascular Disease: An Update from Molecular Mechanism to Clinical Perspective. Front. Pharmacol. 2020, 11, 582680.
- Wang, M.; Wu, J.; Jiao, H.; Oluwabiyi, C.; Li, H.; Zhao, J.; Zhou, Y.; Wang, X.; Lin, H. Enterocyte synthesizes and secrets uric acid as antioxidant to protect against oxidative stress via the involvement of Nrf pathway. Free Radic. Biol. Med. 2022, 179, 95–108.
- Liu, N.; Xu, H.; Sun, Q.; Yu, X.; Chen, W.; Wei, H.; Jiang, J.; Xu, Y.; Lu, W. The Role of Oxidative Stress in Hyperuricemia and Xanthine Oxidoreductase (XOR) Inhibitors. Oxid. Med. Cell. Longev. 2021, 2021, 1470380.
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553.
- Liu, M.; Sun, X.; Chen, B.; Dai, R.; Xi, Z.; Xu, H. Insights into Manganese Superoxide Dismutase and Human Diseases. Int. J. Mol. Sci. 2022, 23, 5893.
- Banks, C.J.; Andersen, J.L. Mechanisms of SOD1 regulation by post-translational modifications. Redox Biol. 2019, 26, 101270.
- Coates, L.; Sullivan, B. The macromolecular neutron diffractometer at the spallation neutron source. Methods Enzymol. 2020, 634, 87–99.
- Tak, L.J.; Kim, H.Y.; Ham, W.K.; Agrahari, G.; Seo, Y.; Yang, J.W.; An, E.J.; Bang, C.H.; Lee, M.J.; Kim, H.S.; et al. Superoxide Dismutase 3-Transduced Mesenchymal Stem Cells Preserve Epithelial Tight Junction Barrier in Murine Colitis and Attenuate Inflammatory Damage in Epithelial Organoids. Int. J. Mol. Sci. 2021, 22, 6431.
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167.
- Zandi, P.; Schnug, E. Reactive Oxygen Species, Antioxidant Responses and Implications from a Microbial Modulation Perspective. Biology 2022, 11, 155.
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108.
- Yang, Z.; Huang, Y.; Zhu, L.; Yang, K.; Liang, K.; Tan, J.; Yu, B. SIRT6 promotes angiogenesis and hemorrhage of carotid plaque via regulating HIF-1alpha and reactive oxygen species. Cell Death Dis. 2021, 12, 77.
- Handy, D.E.; Loscalzo, J. The role of glutathione peroxidase-1 in health and disease. Free Radic. Biol. Med. 2022, 188, 146–161.
- Zhang, J.; Hao, H.; Wu, X.; Wang, Q.; Chen, M.; Feng, Z.; Chen, H. The functions of glutathione peroxidase in ROS homeostasis and fruiting body development in Hypsizygus marmoreus. Appl. Microbiol. Biotechnol. 2020, 104, 10555–10570.
- Prasai, P.K.; Shrestha, B.; Orr, A.W.; Pattillo, C.B. Decreases in GSH:GSSG activate vascular endothelial growth factor receptor 2 (VEGFR2) in human aortic endothelial cells. Redox Biol. 2018, 19, 22–27.
- Papachristoforou, E.; Lambadiari, V.; Maratou, E.; Makrilakis, K. Association of Glycemic Indices (Hyperglycemia, Glucose Variability, and Hypoglycemia) with Oxidative Stress and Diabetic Complications. J. Diabetes Res. 2020, 2020, 7489795.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
781
Revisions:
2 times
(View History)
Update Date:
07 Jun 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No