Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Jiemei Wang and Version 2 by Conner Chen.
Reactive oxygen species (ROS) are radical oxygen intermediates that serve as important second messengers in signal transduction. However, when the accumulation of these molecules exceeds the buffering capacity of antioxidant enzymes, oxidative stress and endothelial cell (EC) dysfunction occur.
- endothelial cells
- oxidative stress
- ROS
1. Introduction
Reactive oxygen species (ROS) are byproducts of enzymatic reactions, composed of both free radical and non-free radical oxygen intermediates, and are generated in numerous cell compartments such as the endoplasmic reticulum (ER), mitochondria, and cell membrane [1][2][1,2]. At physiological concentrations, ROS are generated by several sources, including uncoupled endothelial nitric oxide synthases (eNOS), NADPH oxidases (NOX), xanthine oxidases (XO), cyclooxygenases, and the mitochondrial electron transport chain [3]. Antioxidant enzymes such as superoxide dismutase (SOD), catalase (CAT), and peroxidase (POD) are required to manage the production of ROS for cellular health [4][5][4,5].
Cardiovascular (CV) diseases, the leading causes of death worldwide, act to remodel blood vessels and restrict blood flow to the heart and nervous system [6]. Oxidative stress contributes to the development of CV risks and disorders, including hypertension, atherosclerosis, diabetes mellitus (DM), cardiomyopathy, obesity, and congestive heart failure [7][8][7,8]. The endothelium not only regulates the passage of nutrients between tissues, forms the inner lining of all blood vessels, and manages the protective barrier properties of the vascular system [9][10][9,10], but also modulates vascular growth and permeability, tissue metabolism, immune responses to inflammatory stimulation, stem cell recruitment, and regulation of vascular tone [11][12][11,12]. Endothelial cells (ECs) also secrete a wide range of cytokines and growth factors that regulate various physiological activities in autocrine, paracrine, and endocrine manners. One of the important molecules that ECs produce is nitric oxide (NO), generated by endothelial NO synthase (eNOS). NO is a vasoactive molecule that participates in vascular remodeling, vasodilation, platelet aggregation and adhesion, clot formation, and renal hemodynamics [13]. ROS serve as important second messengers for signal transduction and aid in modulating EC activation, proliferation, and angiogenesis [14][15][14,15]. However, under CV risks or disorders when the accumulation of ROS exceeds the buffering capacity of antioxidant enzymes, oxidative stress and molecular damage occur, leading to early apoptotic death and genomic alteration in ECs [16]. ECs then switch from a vasodilative, anti-inflammatory, anticoagulant environment to a vasoconstrictive, proinflammatory, and procoagulant environment, with decreased bioavailability of nitric oxide (NO) and overproduction of superoxide (O2−), leading to impaired cellular repair [17][18][17,18] and endothelial dysfunction.
Extensive studies have investigated the therapeutic potential of various antioxidant therapies in ECs [6], with recent studies turning to the investigation of microRNAs (miRNA). MicroRNAs are short, non-coding RNA molecules that silence post-transcriptional target genes by promoting messenger RNA (mRNA) cleavage and/or inhibition of protein translation [19][20][19,20]. Emerging evidence has suggested that ROS and microRNAs co-regulate each other. ROS dysregulate the expression of miRNAs in vascular and immune cells, while miRNA manages oxidative stress by targeting specific mRNAs [21]. Due to the biodistribution of these molecules and the presence of multiple mRNA targets for a single microRNA, mechanistic approaches for reducing excessive oxidative stress without impacting other pathways have been evolving [22].
2. Generation of ROS in Endothelial Cells
ECs actively respond to hemodynamic changes and blood-borne signals and facilitate the exchange of oxygen, nutrients, solutes, hormones, and macromolecules between the blood and surrounding tissues, making them essential in the management of metabolic homeostasis [23]. Production of ROS by ECs is a required process for normal cellular function and can be triggered by oxidants produced by activated immune cells, cytokines, and other physical stimuli [23]. Intracellular ROS consist of superoxide, hydrogen peroxide (H2O2), and hydroxyl radicals that are derived from uncoupled eNOS, NADPH oxidase, electron transport chain in mitochondria, and xanthine oxidase (Figure 1) [24][25][24,25]. Under controlled environments, these ROS serve as second messengers regulating cell proliferation, differentiation, immune responses, and tissue repair [26]. However, an overabundance of ROS leads to EC dysfunction, increasing the risk of atherosclerosis, hypertension, hypercholesterolemia, stroke, diabetes, obesity, and other metabolic syndromes [27][28][27,28].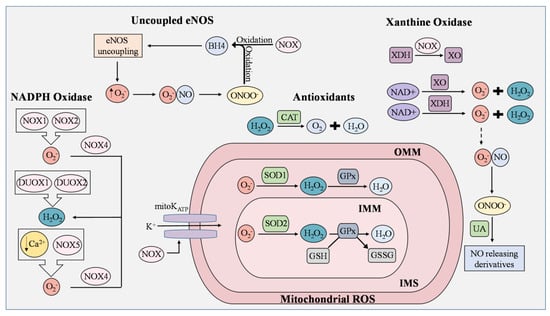
Figure 1. ROS generation and removal in endothelial cells. Reactive oxygen species (ROS) are mainly derived from uncoupled endothelial nitric oxide synthase (eNOS), NADPH oxidase (NOX), mitochondria, and xanthine oxidase (XO). Antioxidants that regulate ROS include catalase (CAT), superoxide dismutase 1 (SOD1), superoxide dismutase 2 (SOD2), and uric acid (UA). During eNOS uncoupling, superoxide (O2−) is produced in abundance rather than nitric oxide (NO). Superoxide then reacts with NO to generate peroxynitrite (ONOO−), which rapidly oxidizes tetrahydrobiopterin (BH4), thereby maintaining the state of eNOS uncoupling. ROS produced from NOX isoforms are involved in proliferation, migration, and differentiation in ECs, and are known to cause mitochondrial DNA damage, induce oxidative inactivation of BH4, stimulate the conversion of xanthine dehydrogenase (XDH) to XO, and manipulate the opening of the mitochondrial ATP-sensitive K+ ion channel (mitoKATP). NOX1 and NOX2 generate superoxide as opposed to NOX4, which converts superoxide to H2O2. In low concentrations of Ca2+, NOX5 enhances ROS production in ECs. Dual oxidase 1 (DUOX1) and dual oxidase 2 (DUOX2) possess roles in immune response and cell differentiation and are responsible for H2O2 generation. Leakage of electrons from the mitochondrial electron transport chain generates superoxide, but the presence of SOD1 and SOD2 prevents the buildup of damaging mitochondrial ROS (mROS). In the inner mitochondrial matrix (IMM), superoxide is converted to H2O2 by SOD2, while superoxide in the intermembrane space (IMS), which is between the IMM and outer mitochondrial matrix (OMM), is converted by SOD1. H2O2 is reduced to H2O by oxidizing glutathione (GSH) into glutathione disulfide (GSSG). This reaction is catalyzed by glutathione peroxidase (GPx). Catalase is an Fe-containing enzyme that catalyzes hydrogen peroxide into H2O and molecular oxygen. XO and XDH are the two interconvertible forms of xanthine oxidoreductase (XOR) where NAD+ is used to generate superoxide and peroxide. Under normal physiological conditions, UA possesses antioxidant activity and can aid in the regulation of ROS generated by XOR by reacting with ONOO− to form NO-releasing derivatives.