 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pedro Sousa Sampaio | -- | 2193 | 2023-06-06 09:59:38 | | | |
| 2 | Catherine Yang | Meta information modification | 2193 | 2023-06-06 10:08:29 | | |
Video Upload Options
Biocatalysis is currently a workhorse used to produce a wide array of compounds, from bulk to fine chemicals, in a green and sustainable manner. The success of biocatalysis is largely thanks to an enlargement of the feasible chemical reaction toolbox. This materialized due to major advances in enzyme screening tools and methods, together with high-throughput laboratory techniques for biocatalyst optimization through enzyme engineering. Therefore, enzyme-related knowledge has significantly increased. To handle the large number of data now available, computational approaches have been gaining relevance in biocatalysis, among them machine learning methods (MLMs). MLMs use data and algorithms to learn and improve from experience automatically.
1. An Overview and Basic Concepts
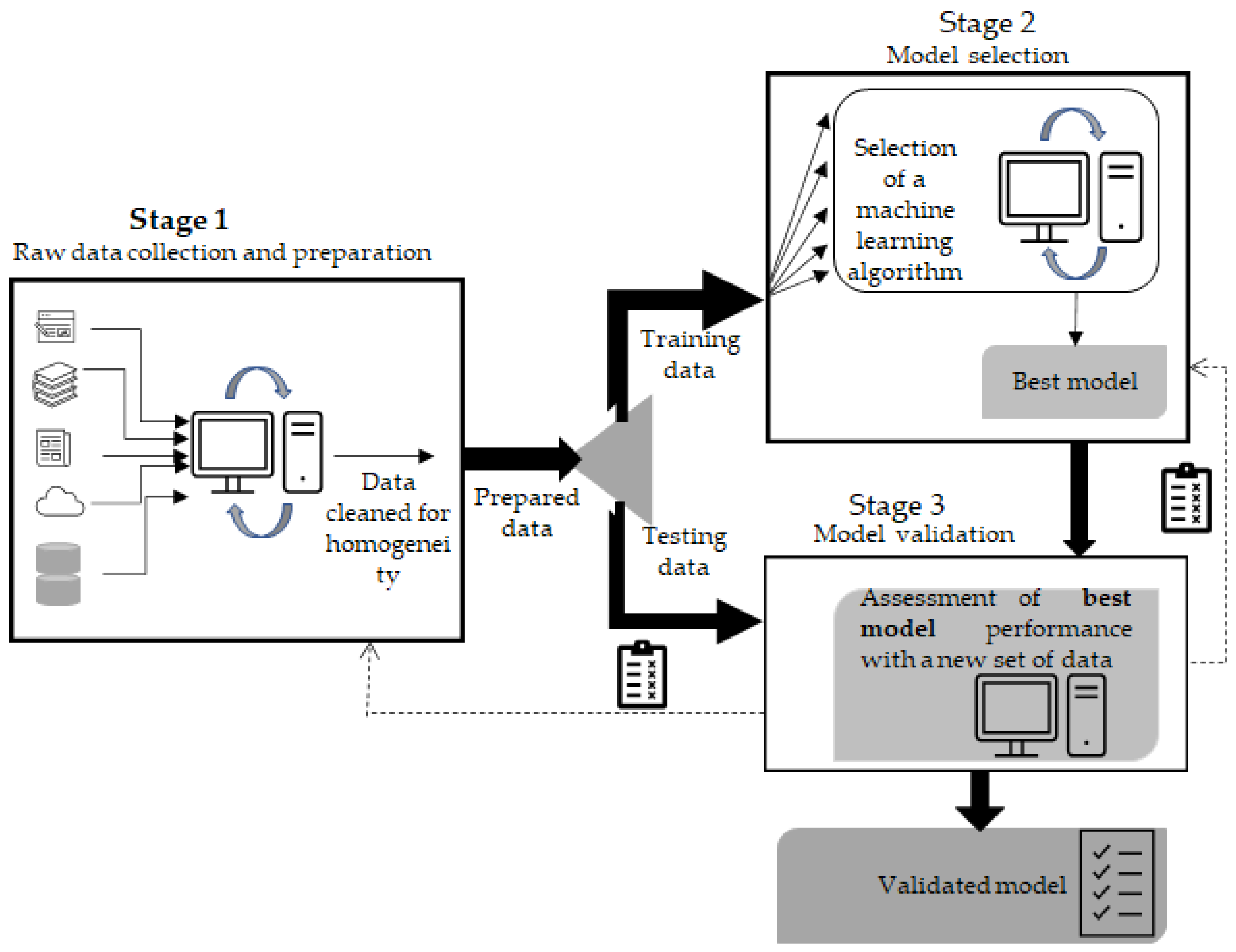
2. Building a Machine Learning Sequence–Function Model
3. ML Algorithms
References
- Jang, W.D.; Kim, G.B.; Kim, Y.; Lee, S.Y. Applications of Artificial Intelligence to Enzyme and Pathway Design for Metabolic Engineering. Curr. Opin. Biotechnol. 2022, 73, 101–107.
- Ferruz, N.; Schmidt, S.; Höcker, B. ProtGPT2 Is a Deep Unsupervised Language Model for Protein Design. Nat. Commun. 2022, 13, 4348.
- Villalobos-Alva, J.; Ochoa-Toledo, L.; Villalobos-Alva, M.J.; Aliseda, A.; Pérez-Escamirosa, F.; Altamirano-Bustamante, N.F.; Ochoa-Fernández, F.; Zamora-Solís, R.; Villalobos-Alva, S.; Revilla-Monsalve, C.; et al. Protein Science Meets Artificial Intelligence: A Systematic Review and a Biochemical Meta-Analysis of an Inter-Field. Front. Bioeng. Biotechnol. 2022, 10, 788300.
- Pan, X.; Kortemme, T. Recent Advances in de Novo Protein Design: Principles, Methods, and Applications. J. Biol. Chem. 2021, 296, 100558.
- Singh, N.; Malik, S.; Gupta, A.; Srivastava, K.R. Revolutionizing Enzyme Engineering through Artificial Intelligence and Machine Learning. Emerg Top Life Sci. 2021, 5, 113–125.
- Cadet, X.F.; Gelly, J.C.; van Noord, A.; Cadet, F.; Acevedo-Rocha, C.G. Learning Strategies in Protein Directed Evolution. In Directed Evolution: Methods and Protocols; Currin, A., Swainston, N., Eds.; Springer US: New York, NY, USA, 2022; pp. 225–275. ISBN 978-1-0716-2152-3.
- Saito, Y.; Oikawa, M.; Sato, T.; Nakazawa, H.; Ito, T.; Kameda, T.; Tsuda, K.; Umetsu, M. Machine-Learning-Guided Library Design Cycle for Directed Evolution of Enzymes: The Effects of Training Data Composition on Sequence Space Exploration. ACS Catal. 2021, 11, 14615–14624.
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the Sequence Specificities of DNA- and RNA-Binding Proteins by Deep Learning. Nat. Biotechnol. 2015, 33, 831–838.
- Hui, S.; Xing, X.; Bader, G.D. Predicting PDZ Domain Mediated Protein Interactions from Structure. BMC Bioinform. 2013, 14, 27.
- Poplin, R.; Chang, P.-C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A Universal SNP and Small-Indel Variant Caller Using Deep Neural Networks. Nat. Biotechnol. 2018, 36, 983–987.
- Dias-Audibert, F.L.; Navarro, L.C.; de Oliveira, D.N.; Delafiori, J.; Melo, C.F.O.R.; Guerreiro, T.M.; Rosa, F.T.; Petenuci, D.L.; Watanabe, M.A.E.; Velloso, L.A.; et al. Combining Machine Learning and Metabolomics to Identify Weight Gain Biomarkers. Front. Bioeng. Biotechnol. 2020, 8, 6.
- Erban, A.; Fehrle, I.; Martinez-Seidel, F.; Brigante, F.; Más, A.L.; Baroni, V.; Wunderlin, D.; Kopka, J. Discovery of Food Identity Markers by Metabolomics and Machine Learning Technology. Sci. Rep. 2019, 9, 9697.
- Ghaffari, M.H.; Jahanbekam, A.; Sadri, H.; Schuh, K.; Dusel, G.; Prehn, C.; Adamski, J.; Koch, C.; Sauerwein, H. Metabolomics Meets Machine Learning: Longitudinal Metabolite Profiling in Serum of Normal versus Overconditioned Cows and Pathway Analysis. J. Dairy Sci. 2019, 102, 11561–11585.
- Liebal, U.W.; Phan, A.N.T.; Sudhakar, M.; Raman, K.; Blank, L.M. Machine Learning Applications for Mass Spectrometry-Based Metabolomics. Metabolites 2020, 10, 243.
- Heinemann, D. (Ed.) Praxiskommentar Transparenzgesetz (LTranspG RLP), 1st ed.; Springer Fachmedien Wiesbaden: Wiesbaden, Germany, 2019; ISBN 978-3-658-18436-0.
- Helmy, M.; Smith, D.; Selvarajoo, K. Systems Biology Approaches Integrated with Artificial Intelligence for Optimized Metabolic Engineering. Metab. Eng. Commun. 2020, 11, e00149.
- Cuperlovic-Culf, M. Machine Learning Methods for Analysis of Metabolic Data and Metabolic Pathway Modeling. Metabolites 2018, 8, 4.
- Mazurenko, S.; Prokop, Z.; Damborsky, J. Machine Learning in Enzyme Engineering. ACS Catal. 2020, 10, 1210–1223.
- Yan, B.; Ran, X.; Gollu, A.; Cheng, Z.; Zhou, X.; Chen, Y.; Yang, Z.J. IntEnzyDB: An Integrated Structure–Kinetics Enzymology Database. J. Chem. Inf. Model. 2022, 62, 5841–5848.
- Pleiss, J. Standardized Data, Scalable Documentation, Sustainable Storage—EnzymeML as A Basis for FAIR Data Management In Biocatalysis. ChemCatChem 2021, 13, 3909–3913.
- Minkiewicz, P.; Darewicz, M.; Iwaniak, A.; Bucholska, J.; Starowicz, P.; Czyrko, E. Internet Databases of the Properties, Enzymatic Reactions, and Metabolism of Small Molecules—Search Options and Applications in Food Science. Int. J. Mol. Sci. 2016, 17, 2039.
- Chicco, D.; Oneto, L.; Tavazzi, E. Eleven Quick Tips for Data Cleaning and Feature Engineering. PLoS Comput. Biol. 2022, 18, e1010718.
- Menke, M.J.; Behr, A.S.; Rosenthal, K.; Linke, D.; Kockmann, N.; Bornscheuer, U.T.; Dörr, M. Development of an Ontology for Biocatalysis. Chem. Ing. Tech. 2022, 94, 1827–1835.
- Bur, A.M.; Shew, M.; New, J. Artificial Intelligence for the Otolaryngologist: A State of the Art Review. Otolaryngol. Head Neck Surg. 2019, 160, 603–611.
- Niroula, A.; Vihinen, M. Variation Interpretation Predictors: Principles, Types, Performance, and Choice. Hum. Mutat. 2016, 37, 579–597.
- Sharma, A.; Mishra, P.K. State-of-the-Art in Performance Metrics and Future Directions for Data Science Algorithms. J. Sci. Res. 2020, 64, 221–238.
- Badillo, S.; Banfai, B.; Birzele, F.; Davydov, I.I.; Hutchinson, L.; Kam-Thong, T.; Siebourg-Polster, J.; Steiert, B.; Zhang, J.D. An Introduction to Machine Learning. Clin. Pharmacol. Ther. 2020, 107, 871–885.
- Yang, K.K.; Wu, Z.; Arnold, F.H. Machine-Learning-Guided Directed Evolution for Protein Engineering. Nat. Methods 2019, 16, 687–694.
- Cai, Z.; Long, Y.; Shao, L. Classification Complexity Assessment for Hyper-Parameter Optimization. Pattern Recognit. Lett. 2019, 125, 396–403.
- Abbott, A.S.; Turney, J.M.; Zhang, B.; Smith, D.G.A.; Altarawy, D.; Schaefer, H.F. PES-Learn: An Open-Source Software Package for the Automated Generation of Machine Learning Models of Molecular Potential Energy Surfaces. J. Chem. Theory Comput. 2019, 15, 4386–4398.
- Hoopes, A.; Hoffmann, M.; Fischl, B.; Guttag, J.; Dalca, A.V. HyperMorph: Amortized Hyperparameter Learning for Image Registration. In International Conference on Information Processing in Medical Imaging; Springer: Berlin/Heidelberg, Germany, 2021; pp. 3–17.
- Mowbray, M.; Savage, T.; Wu, C.; Song, Z.; Cho, B.A.; Del Rio-Chanona, E.A.; Zhang, D. Machine Learning for Biochemical Engineering: A Review. Biochem. Eng. J. 2021, 172, 108054.
- Basha, S.M.; Rajput, D.S. Survey on Evaluating the Performance of Machine Learning Algorithms: Past Contributions and Future Roadmap. In Deep Learning and Parallel Computing Environment for Bioengineering Systems; Elsevier: Amsterdam, The Netherlands, 2019; pp. 153–164.
- Abraham, G.K.; Jayanthi, V.S.; Bhaskaran, P. Convolutional Neural Network for Biomedical Applications. In Computational Intelligence and Its Applications in Healthcare; Elsevier: Amsterdam, The Netherlands, 2020; pp. 145–156.
- Fox, R.J.; Davis, S.C.; Mundorff, E.C.; Newman, L.M.; Gavrilovic, V.; Ma, S.K.; Chung, L.M.; Ching, C.; Tam, S.; Muley, S.; et al. Improving Catalytic Function by ProSAR-Driven Enzyme Evolution. Nat. Biotechnol. 2007, 25, 338–344.
- Li, Y.; Drummond, D.A.; Sawayama, A.M.; Snow, C.D.; Bloom, J.D.; Arnold, F.H. A Diverse Family of Thermostable Cytochrome P450s Created by Recombination of Stabilizing Fragments. Nat. Biotechnol. 2007, 25, 1051–1056.
- Walsh, I.; Myint, M.; Nguyen-Khuong, T.; Ho, Y.S.; Ng, S.K.; Lakshmanan, M. Harnessing the Potential of Machine Learning for Advancing “Quality by Design” in Biomanufacturing. MAbs 2022, 14, 2013593.
- Helleckes, L.M.; Hemmerich, J.; Wiechert, W.; von Lieres, E.; Grünberger, A. Machine Learning in Bioprocess Development: From Promise to Practice. Trends Biotechnol. 2022, 41, 817–835.
- Mowbray, M.; Vallerio, M.; Perez-Galvan, C.; Zhang, D.; Del Rio Chanona, A.; Navarro-Brull, F.J. Industrial Data Science—A Review of Machine Learning Applications for Chemical and Process Industries. React. Chem. Eng. 2022, 7, 1471–1509.
- Lim, S.J.; Son, M.; Ki, S.J.; Suh, S.-I.; Chung, J. Opportunities and Challenges of Machine Learning in Bioprocesses: Categorization from Different Perspectives and Future Direction. Bioresour. Technol. 2023, 370, 128518.

