Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hyunju Kang | -- | 3492 | 2023-06-06 04:35:54 | | | |
| 2 | Rita Xu | Meta information modification | 3492 | 2023-06-06 04:43:50 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kang, H. Vascular Inflammation and Atherosclerosis. Encyclopedia. Available online: https://encyclopedia.pub/entry/45214 (accessed on 23 July 2026).
Kang H. Vascular Inflammation and Atherosclerosis. Encyclopedia. Available at: https://encyclopedia.pub/entry/45214. Accessed July 23, 2026.
Kang, Hyunju. "Vascular Inflammation and Atherosclerosis" Encyclopedia, https://encyclopedia.pub/entry/45214 (accessed July 23, 2026).
Kang, H. (2023, June 06). Vascular Inflammation and Atherosclerosis. In Encyclopedia. https://encyclopedia.pub/entry/45214
Kang, Hyunju. "Vascular Inflammation and Atherosclerosis." Encyclopedia. Web. 06 June, 2023.
Copy Citation
Atherosclerosis (AS) is a chronic metabolic disorder and the primary cause of cardiovascular diseases, resulting in substantial morbidity and mortality worldwide. Initiated by endothelial cell stimulation, AS is characterized by arterial inflammation, lipid deposition, foam cell formation, and plaque development. Certain nutrients such as carotenoids, polyphenols, and vitamins can prevent the atherosclerotic process by modulating inflammation and metabolic disorders through the regulation of gene acetylation states, mediated with histone deacetylases (HDACs).
nutrients
acetylation state
vascular inflammation
atherosclerosis
1. Introduction
Atherosclerosis (AS), a chronic inflammatory ailment of the arterial system, pertains to the intima of medium or large arteries, associated with dyslipidemia as well as alterations in the arterial wall composition [1]. The inflammatory process leads to vascular dysfunction by forming atherosclerotic plaques due to vascular inflammation (VI). The initial pathogenic event in AS is the dysfunction of endothelial cells (ECs), resulting from the disturbed vascular flow. The excessive production of reactive oxygen species (ROS), by enzymatic pathways including uncoupled nitric oxide synthase (NOS) and NADPH oxidases (NOXs), leads to dysfunction of ECs and vascular smooth muscle cells (VSMCs), and inflammation, leading to AS. AS has been accompanied by accumulated modified lipids, foam cell formation, vascular wall fibroblasts, apoptosis, and calcification [2].
Epigenetic therapy, mediated by histone modifications, has proven effective in regulating acetylation states through nutrient administration. Pharmacological modulation of acetylation states by activating class III histone deacetylases (HDACs), such as sirtuin 1 and sirtuin 3 (SIRT1 and SIRT3) with small natural nutrient molecules, has been shown to protect against inflammation and metabolic dysfunction associated with VI and AS [3]. Conversely, inhibition of class I and II HDACs by nutrients has been suggested to ameliorate inflammatory and metabolic responses. Dynamic epigenetic regulation governing differentiation and activation of monocytes and macrophages, may affect inflammatory and oxidative pathways associated with AS [4]. Epigenetic regulation of enzymes that control excessive ROS production by HDACs, such as SIRTs, has been proposed to inhibit the progression of AS by suppressing EC dysfunction and inflammation [3][5]. SIRT1 is known to protect against various oxidative stress and aging-induced EC dysfunction and inflammation. This protective effect is achieved through deacetylation, which subsequently enhances the ability of cellular components to inhibit inflammatory and oxidative responses [6]. Foam cell formation by VSMCs, monocytes, or macrophages can initiate the development of AS [7]. The process of reverse cholesterol transport, from peripheral cells to the liver, can effectively eliminate excess cholesterol from arterial cells, thus preventing foam cell formation from VSMCs and macrophages [8]. Hepatic lipid accumulation can be reduced by SIRT1 by activating AMP-activated protein kinase (AMPK) [9] and peroxisome proliferator-activated receptor α (PPARα) signaling [3].
Long-term epigenetic remodeling of innate immune cells induced by pro-atherogenic stimuli, including low-density lipoprotein (LDL) cholesterol and oxidized LDL, can trigger continuous activation even after the stimuli are removed [10]. In other words, innate immune cells such as monocytes, macrophages, or natural killer cells can acquire non-specific memories by epigenetic modulation that alters their response to subsequent stimuli, based on this trained immune system [11]. Dietary stress induced by alcohol, high triglyceride, or high cholesterol diets has been proven to impede the activation of SIRT1 and SIRT3, which in turn promotes VI and the development of atherosclerotic plaques [12]. Cumulative studies showed the epigenetic control of VI and AS by DNA methylation [4]; however, relatively little attention has been paid to pathways leading to nutrient-related VI and AS by regulating the acetylation state. Given that dietary stress stimulates the histone acetyltransferases (HATs) to enhance the transcription of AS-related genes, regulating HDACs activity through the control of the acetylation state may offer potential roles for nutrients in addressing the pathogenesis leading to VI and AS.
2. Epigenetic Regulation of VI and AS
Epigenetic regulations of histone acetylation state are composed of three steps: detection, acetylation, and deacetylation [13]. Histone acetylation is initiated by detecting specific signaling in the bromodomain and extra-terminal domain family of proteins, called readers, which bind acetyl–lysine and link to a series of covalent histone modifications. Histone acetylation is mediated by HATs called writers, while deacetylation is mediated by HDACs called erasers [14]. The coordinated epigenetic action of readers, writers, and erasers with the help of various related enzymes leads to fine-tuned gene expression by relaxing or condensing chromatin structure to target gene promoters.
2.1. Factors Affecting VI and AS
Vascular ECs facilitate a thin layer in the inner wall of blood vessels to regulate their responses to diverse stimuli, including oxidative stress, inflammation, and shear stress [15]. Endothelial dysfunction, characterized by increased endothelial vascular permeability and the migration and proliferation of ECs, serves as an indicator of AS [2]. Risk factors for AS include hypertension, smoking, hyperlipidemia, insulin resistance, obesity, and metabolic dysfunction [1]. Substantial alterations in blood vessel morphology, such as bifurcation, flexion, and narrowing of arteries, contribute to the increased incidence of AS [2]. Pathophysiological processes of AS are directly associated with VI, which can be influenced by various stresses and dysfunctions, including an imbalance of blood glucose and plasma lipid in the artery [15]. To avoid VI and prevent AS, it is essential to maintain the homeostasis of blood glucose and plasma lipid, which can be regulated by the conditions of vascular ECs. In particular, regulating detrimental events such as inflammation, shear stress, and oxidative stress is a crucial factor for maintaining the homeostasis of blood vessels [16]. Endothelial dysfunctions, resulting in enhanced vascular permeability and the migration and proliferation of ECs, trigger AS [17].
Vascular dysfunction and features of AS can be attributed to both physical and chemical factors. The physical factors are ascribed to the disturbing forces of blood flow in arteries, including fluid shear stress, tensile forces, and hydrostatic pressure forces [18]. Disturbed blood flow in the curvature and bifurcation regions can promote inflammation and AS by activating activator protein 1 (AP-1), nuclear factor-κB (NF-κB), and protein kinase C (PKC) [19]. The chemical risk factors contributing to AS involve inflammation, oxidative stress, and bioavailability of nitric oxide (NO), which plays a regulatory role in managing the redox state, NF-κB, p53, and endothelial NOS (eNOS) pathways [20]. Inflammatory molecules such as interleukin-1 (IL-1), IL-6, cyclooxygenase-2 (COX-2), matrix metalloproteinase-2 (MMP-2), and MMP-9, adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1), and angiogenic growth factors can induce inflammation and EC dysfunction, resulting in vascular remodeling and angiogenesis [10].
From an immunological view, inflammatory molecules play a crucial role in recruiting and promoting monocytes to transmigrate across the endothelial monolayer into the sub-intima of the vessel wall, where they proliferate and differentiate into macrophages and foam cells by taking up lipoproteins [21]. The progressive death of these foam cells and macrophages, along with the release of lipid-filled contents and tissue factors lead to the formation of a lipid-rich necrotic core, which destabilizes plaques [21]. Meanwhile, VSMCs migrate from the medial layer and accumulate in the intima, where they form the fibrous cap over the lesion by secreting interstitial collagen and elastin. However, cytokines and matrix-degrading proteases secreted by macrophages and lymphocytes, such as collagenase, gelatinase, stromelysin, and cathepsin, weaken the atherosclerotic plaques [22], eventually breaking the thin fibrous caps and plaques. The exposed pro-coagulant materials released into the blood trigger thrombosis, which can impede blood flow and result in acute stenosis of the arteries [23].
The key features of AS have been identified as a major contributor to cardiovascular disease, increasing the risk of acute myocardial infarction and stroke. Atherosclerotic lesions develop in the intima of arteries due to chronic inflammation, resulting from lipid accumulation with deposited macrophages and T cells. This process leads to increased atherosclerotic plaque growth and triggers blood clot formation [24]. Long-term vascular damage with a chronic inflammatory response, resulting from factors such as high shear stress, free radicals, elevated cholesterol levels, and oxidized LDL, contributes to the initiation of atherosclerotic plaque formation [25].
2.2. Regulation of VI and AS
It has been demonstrated that AS is an epigenetic disorder [14], indicating that epigenetic regulation can be a promising approach to prevent AS [22]. Dietary stress leads to metabolic disorders, such as dysfunctions in glucose and lipid metabolism, which can be ameliorated through epigenetic regulation by utilizing HDACs. The regulation of acetylation states of enzymes and genes involved in oxidative metabolism plays a crucial role in controlling blood glucose and plasma lipid levels, and subsequently, affecting the progression of VI and AS. Figure 1 presents an overview of the epigenetic regulation schemes of nutrients that can inhibit AS.
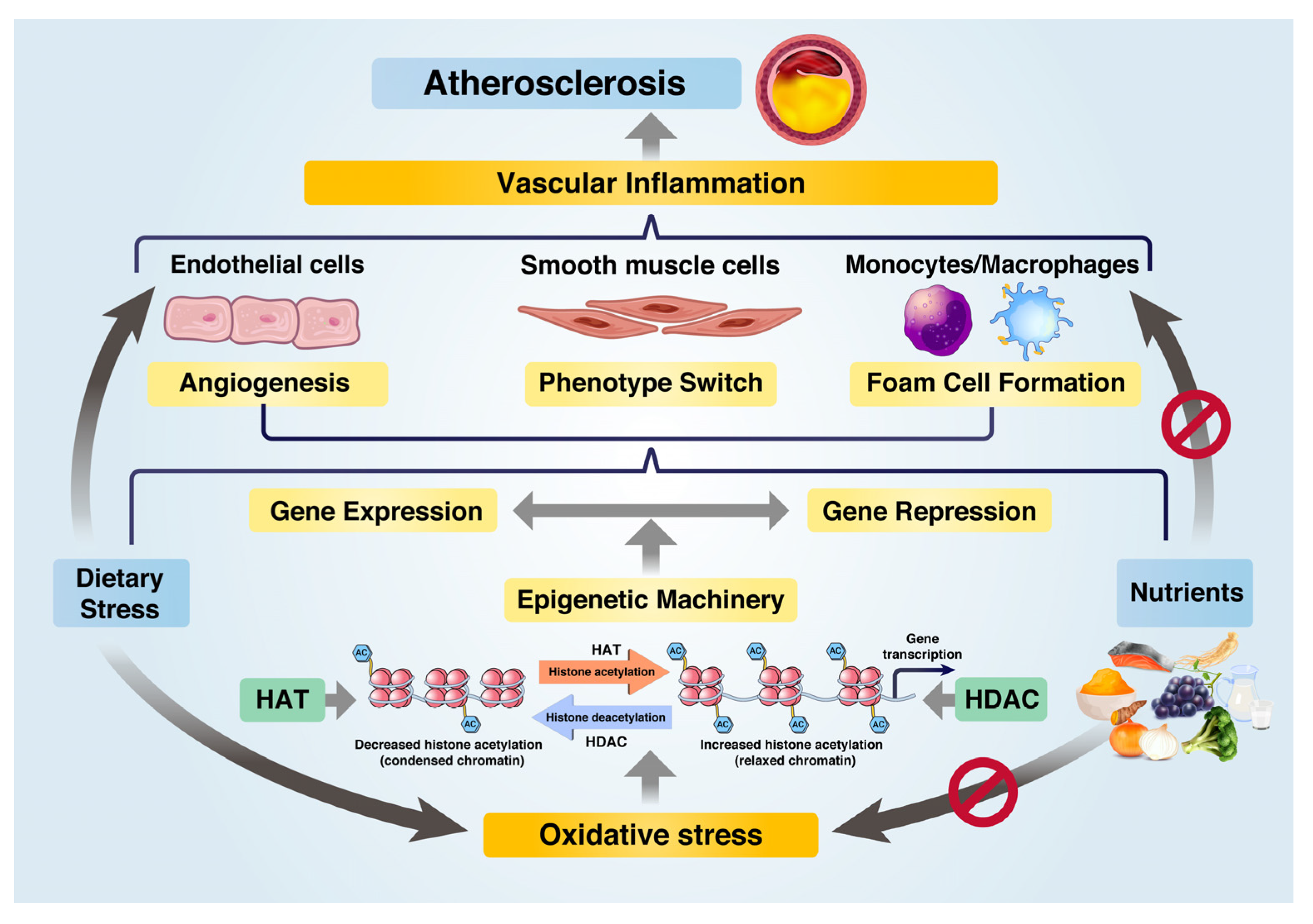
Figure 1. Role of nutrients in preventing atherosclerosis progression by epigenetic regulation of gene expression under dietary stress. Under dietary stress, HATs induce oxidative stress and inflammatory gene expression via epigenetic machinery. Nutrients serve as epigenetic regulators, reducing oxidative stress and repressing inflammatory gene expression through HDACs such as SIRT1 and SIRT3. Nutrients inhibit dietary-induced inflammation and metabolic dysfunctions in ECs, VSMCs, and macrophages, thereby preventing atherosclerosis progression.
Evidence suggests that class II HDACs may contribute to the progression of AS, but SIRT1 has a preventive role in it [3]. SIRT1 can inhibit AS-related chemical risk factors, including inflammation, oxidative stress, and NO bioavailability, by controlling the redox state, inhibiting NF-κB and p53, and activating eNOS [26]. During the initial stages of AS, fat-driven deposits at the vascular walls induce neointima formation by hypercholesterolemia, resulting in hypoxia and induction of hypoxia-inducible factor (HIF)-1, stimulating neovascularization and developing atherosclerotic plaque [27]. SIRT1 can inhibit neointima formation by suppressing HIF-1α expression through deacetylation in hypoxic VSMCs [28]. The structure and function of the arterial wall is determined by the phenotypes of VSMCs, such as their contractile and dedifferentiated state, which play a crucial role in response to atherosclerotic factors. The transition from a contractile to a dedifferentiated state has been shown to have protective effects against AS [29]. During the progression of AS, VSMCs with excessive cholesterol migrate into the intima, where they become modified to an inflammatory phenotype and become the predominant cell type in early arterial intimal thickening [30]. As VSMCs are an essential source of foam cells along with macrophages [31], the epigenetic restoration of VSMC functions reliant on SIRT1 may effectively hinder AS development.
The properties of SIRT1 as an anti-AS can be attributed to its capacity to modulate inflammatory responses and cholesterol metabolism. Specifically, SIRT1 has been shown to suppress the expression of pro-inflammatory mediators such as tumor necrosis factor α (TNFα), IL-6, NF-κB, monocyte chemoattractant protein-1 (MCP-1), ICAM-1, and VCAM-1. Furthermore, it can reduce levels of serum free fatty acids, blood glucose, triglyceride, and total cholesterol, and inhibit inflammatory cell infiltration in atherosclerotic plaques [29]. Given the ability of SIRT1 to regulate common pathways leading to both AS and metabolic disease, it is suggested that the inhibition of AS may be achieved through similar mechanisms employed in metabolic disease prevention, partly attributed to SIRT1 [32].
Foam cell formation can trigger the progression of AS, but this can be prevented by promoting reverse cholesterol transport to remove excess cholesterol from arterial cells, thereby inhibiting foam cell formation in VSMCs and macrophages [8]. The liver can discard accumulated lipids by increasing fatty acid oxidation and ketogenesis, inhibiting hepatic steatosis and inflammation induced by high-fat diets through SIRT1 activation [33]. By suppressing the NF-κB signaling pathway, SIRT1 can reduce the expression of lectin-like oxidized LDL receptor-1 (LOX-1), thereby inhibiting the formation of foam cells in macrophages and monocytes [34]. However, dietary stress induced by alcohol or high-triglyceride or high-cholesterol diet can inhibit the abilities of SIRT1, preventing reverse cholesterol transport and enhancing inflammation, which reduces the deletion of foam cells from atherosclerotic plaques [35].
SIRT1 has been shown to control autophagy by generating a molecular complex and by deacetylating proteins involved in the autophagy process, such as autophagy related 5 (ATG5), ATG7, and ATG8 [36]. This SIRT1-mediated autophagy has been found to protect against foam cell formation in VSMCs treated with oxidized LDL [37]. Furthermore, the toxic effects of oxidized LDL can induce cell apoptosis in atherosclerotic areas, including human smooth muscle cells [38]. The accumulation of apoptotic cells in the vascular wall can lead to plaque erosion, promoting local platelet aggregation and thrombosis, which in turn can prevent the progression of AS [39]. Thus, autophagy appears to be another mechanism by which SIRT1 exerts its anti-atherosclerotic effects.
3. Roles of Nutrients to Inhibit VI and AS
The pathological pathways leading to VI and AS have primarily been attributed to the imbalance in redox and acetylation states, depending on the micro-environmental conditions. The ability of nutrients to inhibit VI and AS can be categorized into two main aspects: their involvement in managing oxidative stress that leads to inflammation and their function in altering histone acetylation states. Core histone proteins, such as histones H3 and H4, are targeted for acetylation by HATs or deacetylation by HDACs [40]. Epigenetic transcription or repression of genes resulting from altered histone acetylation states depends on the modification site of the residues and chromatin remodeling features, which regulate chromatin status as either condensed or relaxed [41]. Dietary stress is known to enhance the expression of inflammation-associated genes through HATs, such as p300 and cAMP-response element binding protein (CBP), while nutrient-derived HDACs exhibit opposite effects [42]. In particular, SIRTs such as SIRT1 and SIRT3 are crucial HDACs induced by various nutrients that participate in essential biological processes through the deacetylation of histones at H3K9, H3K18, and H3K56 [43]. In addition, they deacetylate non-histone protein substrates associated with regulating inflammation and metabolism, such as p53, liver X receptor (LXR), PGC-1α, NF-κB, and forkhead box O protein (FOXO) [43]. The role of nutrients in preventing AS by regulating the balance of redox and acetylation states can be summarized in Figure 2.
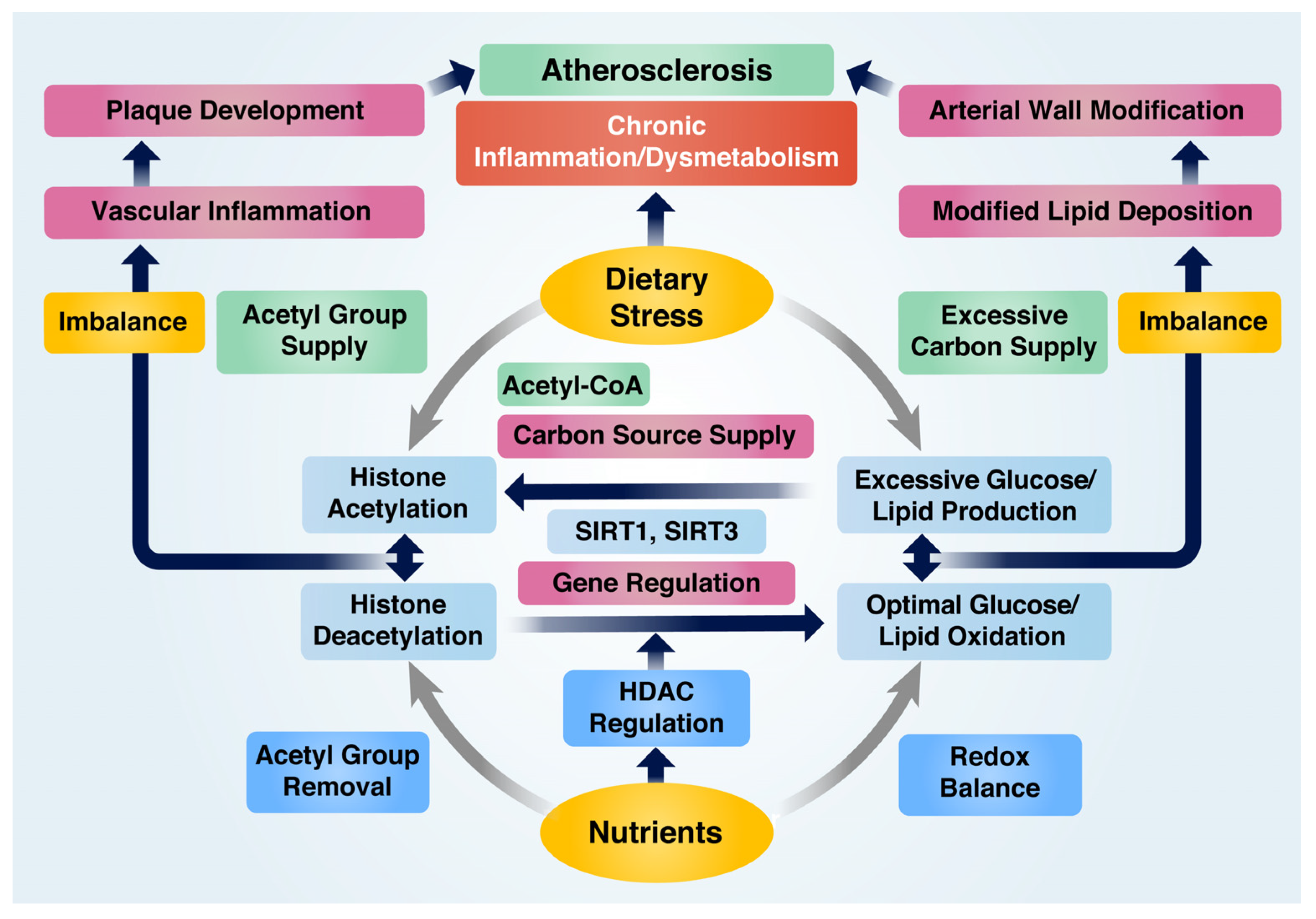
Figure 2. Nutrient-mediated prevention of atherosclerosis by modulating redox and acetylation balance under inflammatory and abnormal metabolic conditions. Dietary stress contributes to histone acetylation by supplying acetyl groups, whereas nutrients facilitate deacetylation. An imbalance between these processes triggers pro-inflammatory gene expression and chronic inflammation. Dietary stress generates an excessive carbon source for the production of extra glucose and lipids, leading to an increased carbon supply for histone acetylation and resulting in hyperglycemia and hyperlipidemia. On the other hand, nutrients regulate genes associated with glucose and lipid oxidation and maintain redox balance. Disruptions in glucose and lipid metabolism cause chronic metabolic dysfunction, but nutrients can modulate chronic inflammation and metabolic dysfunction to prevent atherosclerosis.
3.1. Roles of Nutrients in Regulating Inflammatory Responses
Inflammation, a key characteristic of the innate immune response, can be heightened by the production and translocation of gut-derived lipopolysaccharides (LPS) into the bloodstream. This process triggers the activation of toll-like receptor 4 (TLR4) and NF-κB, which in turn enhances the release of pro-inflammatory cytokines [44]. The production of these cytokines, along with the activation of mitogen-activated protein kinase (MAPK) cascades, such as extracellular signal-regulated kinases (ERKs), and c-Jun N-terminal kinases (JNKs) is initiated by the transforming growth factor β-activated kinase 1 (TAK1) [45]. TLRs activate these signaling pathways by recruiting suitable adaptors, stimulating the generation of pro-inflammatory cytokines. These inflammatory cascades have been closely associated with AS progression, as pattern recognition receptors, such as TLR4, recognize LDLs [44][46]. Pro-inflammatory M1 macrophages play a crucial role in the progression of AS [47], as indicated by their increased presence in atherosclerotic plaques. This suggests that anti-inflammatory agents could be targeted for therapy. The expression of T helper cell 1 (Th1) cytokines, such as TNFα, IL-6, and IL-8, is attributed to the differentiation of M1 macrophages [48]. Additionally, activated platelets release platelet factor 4 (PF4), inducing monocyte differentiation and increasing oxidized LDL uptake by macrophages, which in turn facilitates foam cell formation [49]. These findings support the idea that AS progression can be mitigated by shifting the macrophage phenotype from M1 to M2 [50].
Nutrients play a critical role in preventing inflammatory signaling pathways by effectively inhibiting pro-inflammatory cytokines and blocking downstream inflammatory routes and cascades. The selective functions of nutrients in regulating inflammation stem from their ability to activate SIRTs, such as SIRT1 and SIRT3, while inhibiting classical HDACs [3]. For instance, astaxanthin, xanthophyll carotenoid, significantly decreases the expression of pro-inflammatory genes such as IL-1β, IL-6, as well as TNFα secretion and cytosolic and nuclear NF-κB p65 levels in macrophages. This effect is achieved by preventing inflammation through SIRT1 activation [51]. Additionally, astaxanthin inhibits HDAC4, a class IIa HDAC, which is involved in promoting inflammation due to excessive alcohol [52]. Dietary stress is known to generate an abundance of electrons, ROS, and reactive nitrogen species (RNS), creating an oxidative environment that triggers inflammation [42]. Nutrient-associated enzymes, such as SIRT3, a class III HDAC, have been reported to activate antioxidant genes such as manganese SOD2 and catalase through FOXO3 deacetylation, thereby reducing ROS levels [53]. Evidence suggests that nutrients, including resveratrol and ginsenosides, prevent the development of vascular diseases and reduce the risk of AS by regulating EC metabolism, lipid metabolism, and angiogenesis through SIRT3 activation [54].
Recent studies have demonstrated that the interaction between histone deacetylases, such as SIRT1 and HDAC4, plays a crucial role in regulating inflammation and metabolism [55]. Resveratrol, an activator of SIRT1, impedes gluconeogenesis in insulin-resistant hepatocytes by translocating HDAC4 to the cytoplasm from the nucleus, thereby inactivating it [56]. Moreover, HDAC4 upregulation leads to SIRT1 downregulation during the activation of hepatic stellate cells [57] and in skeletal muscle cells stimulated by interferon γ (IFNγ) [58]. Given that SIRT1 and HDAC4 are involved in modulating inflammation, their interplay has been reported to affect alcohol-induced inflammation in macrophages. For example, astaxanthin inhibits alcohol-triggered inflammation and oxidative stress in macrophages by mediating the opposing actions of SIRT1 and HDAC4 [52]. Selected nutrients, such as resveratrol, activate SIRT3. The deacetylation ability of SIRT3 promotes various antioxidant activities, including SOD2 and catalase. These antioxidants help to maintain the oxidant/antioxidant balance and alleviate vascular endothelial oxidative stress [3].
3.2. Roles of Nutrients in Regulating Histone Acetylation State
Histone acetylation is carried out by HATs, which transfer an acetyl group (CH3CO-) from acetyl-CoA to the lysine-rich histone tails. This process is mediated by bromodomain proteins. In mammalian cells, HATs can be classified into three categories: Gcn5-related N-acetyltransferases (GNAT), MYST, and CBP/p300 [13]. Histone deacetylation, on the other hand, is the reverse process and is carried out by HDACs [22]. Histone acetylation and deacetylation are reversible epigenetic post-translational modifications that are responsive to micro-environmental changes and susceptible to a wide range of modifications [13][22]. As a result, the roles and features of these histone modifications can be employed and utilized by dietary nutrients to regulate inflammatory dysfunctions, such as VI and AS. It has been understood that HATs can be activated by dietary stress stemming from high-fat, high-glycemic diets, or excessive alcohol consumption, as this stress stimulates genes and enzymes sensitive to redox sensing [42]. Nutrients can activate or repress the activity of HDACs through complex mechanisms that depend on their unique structures, functions, and localizations.
Acetyl CoA plays a crucial role in both histone acetylation and de novo lipid synthesis, demonstrating significant effects on histone acetylation and lipid metabolism [59]. Acetyl-CoA can easily enter the nucleus through nuclear pores and acetylate histones, as well as supply acetyl groups for acetylation in the cytoplasm by HAT [60]. Regulating acetyl-CoA levels is essential for controlling the acetylation state of target genes, which in turn affects a series of processes leading to VI and AS. Acetyl-CoA, an acetyl group bound to a cysteine residue of coenzyme A through a thioester bonding, is produced from the conversion of pyruvate by the pyruvate dehydrogenase (PDH) complex in the inner mitochondrial membrane [61]. Excessive ethanol has been reported to significantly decrease PDH activity and inhibit the conversion of pyruvate to acetyl-CoA in macrophages, which leads to an increase in glycolysis rather than the tricarboxylic acid (TCA) cycle. The exceptional increase in glycolysis caused by ethanol can be counteracted by nicotinamide riboside, a nutrient, to restore normal glucose metabolism and energy production [62].
Specific nutrients such as butyrate, ginsenosides, and sulforaphane, known to inhibit class I HDACs such as HDAC1, HDAC2, and HDAC3, may help prevent VI and AS. HDAC1 activation has been associated with a decrease in global H3K9 acetylation in the aortas of apolipoprotein E (ApoE) knockout mice treated with a high methionine diet to induce hyperhomocysteinemia, promoting lipid accumulation in foam cells [63]. HDAC2 overexpression in human aortic ECs suppresses arginase 2 (Arg2) expression, indicating that HDAC2 regulates Arg2 by directly binding to the Arg2 gene promoter. Preventing oxidized LDL-induced HDAC2 downregulation and Arg2 upregulation improves endothelial function [20], suggesting that HDAC2 could be a novel therapy for inhibiting endothelial dysfunction and AS. HDAC3 activates protein kinase B (PKB or Akt) phosphorylation and activity, regulating EC survival during AS development in response to disturbed hemodynamic forces [64]. In macrophages, HDAC3 inhibition promotes an athero-protective phenotype through histone acetylation, accompanied by the gene expression of efflux transporters ATP-binding cassette transporter A1 (ABCA1) and ABCG1, and increases anti-inflammatory and anti-apoptotic capacities [7]. HDAC3 is upregulated in ruptured human atherosclerotic plaques. Furthermore, HDAC3 deletion leads to a pro-fibrotic program via epigenetic regulation of transforming growth factor β1 (TGFβ1), allowing VSMCs to produce collagen and stabilize plaques [65]. This suggests that macrophage specific deletion of HDAC3 could prevent AS development. The efficacy of HDACs in treating VI and AS may depend on the target genes in tissues and organs. For example, HDAC3 deletion in monocytes and macrophages was beneficial for a mouse model associated with AS [66], while blocking HDAC3 decreased cell survival and increased plaque formation in ECs [64]. A single nucleotide polymorphism (rs3791398) in HDAC4 has been reported to associate with carotid intima-media thickness [67].
References
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgozoglu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Prim. 2019, 5, 56.
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid. Med. Cell. Longev. 2019, 2019, 8563845.
- Winnik, S.; Auwerx, J.; Sinclair, D.A.; Matter, C.M. Protective Effects of Sirtuins in Cardiovascular Diseases: From Bench to Bedside. Eur. Heart J. 2015, 36, 3404–3412.
- Xu, S.; Pelisek, J.; Jin, Z.G. Atherosclerosis Is an Epigenetic Disease. Trends Endocrinol. Metab. 2018, 29, 739–742.
- Nicorescu, I.; Dallinga, G.M.; de Winther, M.P.J.; Stroes, E.S.G.; Bahjat, M. Potential Epigenetic Therapeutics for Atherosclerosis Treatment. Atherosclerosis 2019, 281, 189–197.
- Hwang, J.W.; Yao, H.; Caito, S.; Sundar, I.K.; Rahman, I. Redox Regulation of SIRT1 in Inflammation and Cellular Senescence. Free Radic. Biol. Med. 2013, 61, 95–110.
- Van den Bossche, J.; Neele, A.E.; Hoeksema, M.A.; de Heij, F.; Boshuizen, M.C.; van der Velden, S.; de Boer, V.C.; Reedquist, K.A.; de Winther, M.P. Inhibiting Epigenetic Enzymes to Improve Atherogenic Macrophage Functions. Biochem. Biophys. Res. Commun. 2014, 455, 396–402.
- Oram, J.F.; Lawn, R.M. ABCA1. The Gatekeeper for Eliminating Excess Tissue Cholesterol. J. Lipid Res. 2001, 42, 1173–1179.
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. SIRT1 Regulates Hepatocyte Lipid Metabolism through Activating AMP-Activated Protein Kinase. J. Biol. Chem. 2008, 283, 20015–20026.
- Bekkering, S.; Quintin, J.; Joosten, L.A.; van der Meer, J.W.; Netea, M.G.; Riksen, N.P. Oxidized Low-Density Lipoprotein Induces Long-Term Proinflammatory Cytokine Production and Foam Cell Formation via Epigenetic Reprogramming of Monocytes. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1731–1738.
- Bekkering, S.; van den Munckhof, I.; Nielen, T.; Lamfers, E.; Dinarello, C.; Rutten, J.; de Graaf, J.; Joosten, L.A.; Netea, M.G.; Gomes, M.E.; et al. Innate Immune Cell Activation and Epigenetic Remodeling in Symptomatic and Asymptomatic Atherosclerosis in Humans in Vivo. Atherosclerosis 2016, 254, 228–236.
- Stein, S.; Matter, C.M. Protective roles of SIRT1 in atherosclerosis. Cell Cycle 2011, 10, 640–647.
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone Acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120.
- Xu, S.; Kamato, D.; Little, P.J.; Nakagawa, S.; Pelisek, J.; Jin, Z.G. Targeting Epigenetics and Non-Coding RNAs in Atherosclerosis: From Mechanisms to Therapeutics. Pharmacol. Ther. 2019, 196, 15–43.
- Yao, B.C.; Meng, L.B.; Hao, M.L.; Zhang, Y.M.; Gong, T.; Guo, Z.G. Chronic Stress: A Critical Risk Factor for Atherosclerosis. J. Int. Med. Res. 2019, 47, 1429–1440.
- Marin, T.; Gongol, B.; Chen, Z.; Woo, B.; Subramaniam, S.; Chien, S.; Shyy, J.Y.J. Mechanosensitive MicroRNAs—Role in Endothelial Responses to Shear Stress and Redox State. Free Radic. Biol. Med. 2013, 64, 61–68.
- Libby, P. History of Discovery: Inflammation in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2045–2051.
- Gimbrone, M.A.; Anderson, K.R.; Topper, J.N. The Critical Role of Mechanical Forces in Blood Vessel Development, Physiology and Pathology. J. Vasc. Surg. 1999, 29, 1104–1151.
- Nigro, P.; Abe, J.I.; Berk, B.C. Flow Shear Stress and Atherosclerosis: A Matter of Site Specificity. Antioxid. Redox Signal. 2011, 15, 1405–1414.
- Pandey, D.; Hori, D.; Kim, J.H.; Bergman, Y.; Berkowitz, D.E.; Romer, L.H. NEDDylation Promotes Endothelial Dysfunction: A Role for HDAC2. J. Mol. Cell. Cardiol. 2015, 81, 18–22.
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in Atherosclerosis: A Dynamic Balance. Nat. Rev. Immunol. 2013, 13, 709–721.
- Zheng, X.X.; Zhou, T.; Wang, X.A.; Tong, X.H.; Ding, J.W. Histone Deacetylases and Atherosclerosis. Atherosclerosis 2015, 240, 355–366.
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and Challenges in Translating the Biology of Atherosclerosis. Nature 2011, 473, 317–325.
- Neele, A.E.; Van den Bossche, J.; Hoeksema, M.A.; de Winther, M.P. Epigenetic Pathways in Macrophages Emerge as Novel Targets in Atherosclerosis. Eur. J. Pharmacol. 2015, 763, 79–89.
- Hansson, G.K.; Libby, P. The Immune Response in Atherosclerosis: A Double-Edged Sword. Nat. Rev. Immunol. 2006, 6, 508–519.
- Kitada, M.; Ogura, Y.; Koya, D. The Protective Role of Sirt1 in Vascular Tissue: Its Relationship to Vascular Aging and Atherosclerosis. Aging 2016, 8, 2290–2307.
- Doyle, B.; Caplice, N. Plaque Neovascularization and Antiangiogenic Therapy for Atherosclerosis. J. Am. Coll. Cardiol. 2007, 49, 2073–2080.
- Bae, J.U.; Lee, S.J.; Seo, K.W.; Kim, Y.H.; Park, S.Y.; Bae, S.S.; Kim, C.D. SIRT1 Attenuates Neointima Formation by Inhibiting HIF-1alpha Expression in Neointimal Lesion of a Murine Wire-Injured Femoral Artery. Int. J. Cardiol. 2013, 168, 4393–4396.
- Zhang, M.J.; Zhou, Y.; Chen, L.; Wang, X.; Long, C.Y.; Pi, Y.; Gao, C.Y.; Li, J.C.; Zhang, L.L. SIRT1 Improves VSMC Functions in Atherosclerosis. Prog. Biophys. Mol. Biol. 2016, 121, 11–15.
- Orekhov, A.N.; Andreeva, E.R.; Krushinsky, A.V.; Novikov, I.D.; Tertov, V.V.; Nestaiko, G.V.; Khashimov, K.A.; Repin, V.S.; Smirnov, V.N. Intimal Cells and Atherosclerosis. Relationship between the Number of Intimal Cells and Major Manifestations of Atherosclerosis in the Human Aorta. Am. J. Pathol. 1986, 125, 402–415.
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of Intimal Smooth Muscle Cells to Cholesterol Accumulation and Macrophage-like Cells in Human Atherosclerosis. Circulation 2014, 129, 1551–1559.
- Winnik, S.; Stein, S.; Matter, C.M. SIRT1—An Anti-Inflammatory Pathway at the Crossroads between Metabolic Disease and Atherosclerosis. Curr. Vasc. Pharmacol. 2012, 10, 693–696.
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-Specific Deletion of SIRT1 Alters Fatty Acid Metabolism and Results in Hepatic Steatosis and Inflammation. Cell Metab. 2009, 9, 327–338.
- Stein, S.; Lohmann, C.; Schafer, N.; Hofmann, J.; Rohrer, L.; Besler, C.; Rothgiesser, K.M.; Becher, B.; Hottiger, M.O.; Boren, J.; et al. SIRT1 Decreases Lox-1-Mediated Foam Cell Formation in Atherogenesis. Eur. Heart J. 2010, 31, 2301–2309.
- Zeng, H.T.; Fu, Y.C.; Yu, W.; Lin, J.M.; Zhou, L.; Liu, L.; Wang, W. SIRT1 Prevents Atherosclerosis via Liver-X-receptor and NF-kappaB Signaling in a U937 Cell Model. Mol. Med. Rep. 2013, 8, 23–28.
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A Role for the NAD-Dependent Deacetylase Sirt1 in the Regulation of Autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379.
- Li, B.-H.; Yin, Y.-W.; Liu, Y.; Pi, Y.; Guo, L.; Cao, X.-J.; Gao, C.-Y.; Zhang, L.-L.; Li, J.-C. TRPV1 Activation Impedes Foam Cell Formation by Inducing Autophagy in OxLDL-Treated Vascular Smooth Muscle Cells. Cell Death Dis. 2014, 5, e1182.
- Jovinge, S.; Crisby, M.; Thyberg, J.; Nilsson, J. DNA Fragmentation and Ultrastructural Changes of Degenerating Cells in Atherosclerotic Lesions and Smooth Muscle Cells Exposed to Oxidized LDL in Vitro. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2225–2231.
- Mallat, Z.; Tedgui, A. Apoptosis in the Vasculature: Mechanisms and Functional Importance. Br. J. Pharmacol. 2000, 130, 947–962.
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713.
- Bannister, A.J.; Kouzarides, T. Regulation of Chromatin by Histone Modifications. Cell Res. 2011, 21, 381–395.
- Kang, H.; Kim, B. Bioactive Compounds as Inhibitors of Inflammation, Oxidative Stress and Metabolic Dysfunctions via Regulation of Cellular Redox Balance and Histone Acetylation State. Foods 2023, 12, 925.
- Sosnowska, B.; Mazidi, M.; Penson, P.; Gluba-Brzózka, A.; Rysz, J.; Banach, M. The Sirtuin Family Members SIRT1, SIRT3 and SIRT6: Their Role in Vascular Biology and Atherogenesis. Atherosclerosis 2017, 265, 275–282.
- Singh, R.K.; Haka, A.S.; Asmal, A.; Barbosa-Lorenzi, V.C.; Grosheva, I.; Chin, H.F.; Xiong, Y.; Hla, T.; Maxfield, F.R. TLR4 (Toll-Like Receptor 4)-Dependent Signaling Drives Extracellular Catabolism of LDL (Low-Density Lipoprotein) Aggregates. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 86–102.
- Roux, P.P.; Blenis, J. ERK and P38 MAPK-Activated Protein Kinases: A Family of Protein Kinases with Diverse Biological Functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344.
- Howell, K.W.; Meng, X.; Fullerton, D.A.; Jin, C.; Reece, T.B.; Cleveland, J.C., Jr. Toll-like Receptor 4 Mediates Oxidized LDL-Induced Macrophage Differentiation to Foam Cells. J. Surg. Res. 2011, 171, e27–e31.
- Khallou-Laschet, J.; Varthaman, A.; Fornasa, G.; Compain, C.; Gaston, A.T.; Clement, M.; Dussiot, M.; Levillain, O.; Graff-Dubois, S.; Nicoletti, A.; et al. Macrophage Plasticity in Experimental Atherosclerosis. PLoS ONE 2010, 5, e8852.
- de Gaetano, M.; Crean, D.; Barry, M.; Belton, O. M1- and M2-Type Macrophage Responses Are Predictive of Adverse Outcomes in Human Atherosclerosis. Front. Immunol. 2016, 7, 275.
- Nassar, T.; Sachais, B.S.; Akkawi, S.; Kowalska, M.A.; Bdeir, K.; Leitersdorf, E.; Hiss, E.; Ziporen, L.; Aviram, M.; Cines, D.; et al. Platelet Factor 4 Enhances the Binding of Oxidized Low-Density Lipoprotein to Vascular Wall Cells. J. Biol. Chem. 2003, 278, 6187–6193.
- Liu, Y.; Wang, X.; Pang, J.; Zhang, H.; Luo, J.; Qian, X.; Chen, Q.; Ling, W. Attenuation of Atherosclerosis by Protocatechuic Acid via Inhibition of M1 and Promotion of M2 Macrophage Polarization. J. Agric. Food Chem. 2019, 67, 807–818.
- Kang, H.; Lee, Y.; Bae, M.; Park, Y.K.; Lee, J.Y. Astaxanthin Inhibits Alcohol-Induced Inflammation and Oxidative Stress in Macrophages in a Sirtuin 1-Dependent Manner. J. Nutr. Biochem. 2020, 85, 108477.
- Kang, H.; Park, Y.K.; Lee, J.Y. Inhibition of Alcohol-Induced Inflammation and Oxidative Stress by Astaxanthin Is Mediated by Its Opposite Actions in the Regulation of Sirtuin 1 and Histone Deacetylase 4 in Macrophages. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158838.
- Dikalova, A.E.; Itani, H.A.; Nazarewicz, R.R.; McMaster, W.G.; Flynn, C.R.; Uzhachenko, R.; Fessel, J.P.; Gamboa, J.L.; Harrison, D.G.; Dikalov, S.I. Sirt3 Impairment and SOD2 Hyperacetylation in Vascular Oxidative Stress and Hypertension. Circ. Res. 2017, 121, 564–574.
- He, X.; Zeng, H.; Chen, J.X. Emerging Role of SIRT3 in Endothelial Metabolism, Angiogenesis, and Cardiovascular Disease. J. Cell. Physiol. 2019, 234, 2252–2265.
- Kang, H.; Lee, Y.; Kim, M.-B.; Hu, S.; Jang, H.; Park, Y.-K.; Lee, J.-Y. The Loss of Histone Deacetylase 4 in Macrophages Exacerbates Hepatic and Adipose Tissue Inflammation in Male but Not in Female Mice with Diet-Induced Non-Alcoholic Steatohepatitis. J. Pathol. 2021, 255, 319–329.
- Zhao, H.; Shu, L.; Huang, W.; Song, G.; Ma, H. Resveratrol Affects Hepatic Gluconeogenesis via Histone Deacetylase 4. Diabetes Metab. Syndr. Obes. 2019, 12, 401–411.
- Li, M.; Hong, W.; Hao, C.; Li, L.; Xu, H.; Li, P.; Xu, Y. Hepatic Stellate Cell-Specific Deletion of SIRT1 Exacerbates Liver Fibrosis in Mice. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3202–3211.
- Fang, M.; Fan, Z.; Tian, W.; Zhao, Y.; Li, P.; Xu, H.; Zhou, B.; Zhang, L.; Wu, X.; Xu, Y. HDAC4 Mediates IFN-Gamma Induced Disruption of Energy Expenditure-Related Gene Expression by Repressing SIRT1 Transcription in Skeletal Muscle Cells. Biochim. Biophys. Acta 2016, 1859, 294–305.
- He, W.; Li, Q.; Li, X. Acetyl-CoA Regulates Lipid Metabolism and Histone Acetylation Modification in Cancer. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188837.
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl Coenzyme A: A Central Metabolite and Second Messenger. Cell Metab. 2015, 21, 805–821.
- Gray, L.R.; Tompkins, S.C.; Taylor, E.B. Regulation of Pyruvate Metabolism and Human Disease. Cell. Mol. Life Sci. 2014, 71, 2577–2604.
- Kang, H.; Park, Y.K.; Lee, J.Y. Nicotinamide Riboside, an NAD(+) Precursor, Attenuates Inflammation and Oxidative Stress by Activating Sirtuin 1 in Alcohol-Stimulated Macrophages. Lab. Investig. 2021, 101, 1225–1237.
- Zhao, Q.; Li, S.; Li, N.; Yang, X.; Ma, S.; Yang, A.; Zhang, H.; Yang, S.; Mao, C.; Xu, L.; et al. MiR-34a Targets HDAC1-Regulated H3K9 Acetylation on Lipid Accumulation Induced by Homocysteine in Foam Cells. J. Cell. Biochem. 2017, 118, 4617–4627.
- Zampetaki, A.; Zeng, L.; Margariti, A.; Xiao, Q.; Li, H.; Zhang, Z.; Pepe, A.E.; Wang, G.; Habi, O.; Defalco, E.; et al. Histone Deacetylase 3 Is Critical in Endothelial Survival and Atherosclerosis Development in Response to Disturbed Flow. Circulation 2010, 121, 132–142.
- Hoeksema, M.A.; Gijbels, M.J.; Van den Bossche, J.; van der Velden, S.; Sijm, A.; Neele, A.E.; Seijkens, T.; Stoger, J.L.; Meiler, S.; Boshuizen, M.C.; et al. Targeting Macrophage Histone Deacetylase 3 Stabilizes Atherosclerotic Lesions. EMBO Mol. Med. 2014, 6, 1124–1132.
- Zhao, Y.; Mu, H.; Huang, Y.; Li, S.; Wang, Y.; Stetler, R.A.; Bennett, M.V.L.; Dixon, C.E.; Chen, J.; Shi, Y. Microglia-Specific Deletion of Histone Deacetylase 3 Promotes Inflammation Resolution, White Matter Integrity, and Functional Recovery in a Mouse Model of Traumatic Brain Injury. J. Neuroinflamm. 2022, 19, 201.
- Lanktree, M.B.; Hegele, R.A.; Yusuf, S.; Anand, S.S. Multi-Ethnic Genetic Association Study of Carotid Intima-Media Thickness Using a Targeted Cardiovascular SNP Microarray. Stroke 2009, 40, 3173–3179.
More
Information
Subjects:
Pathology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Entry Collection:
Hypertension and Cardiovascular Diseases
Revisions:
2 times
(View History)
Update Date:
06 Jun 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No