Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Juan Segura-Aguilar | -- | 2702 | 2023-05-31 21:20:13 | | | |
| 2 | Rita Xu | Meta information modification | 2702 | 2023-06-01 03:30:00 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Segura-Aguilar, J.; Mannervik, B. Neurodegeneration in Parkinson’s Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/45082 (accessed on 23 July 2026).
Segura-Aguilar J, Mannervik B. Neurodegeneration in Parkinson’s Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/45082. Accessed July 23, 2026.
Segura-Aguilar, Juan, Bengt Mannervik. "Neurodegeneration in Parkinson’s Disease" Encyclopedia, https://encyclopedia.pub/entry/45082 (accessed July 23, 2026).
Segura-Aguilar, J., & Mannervik, B. (2023, May 31). Neurodegeneration in Parkinson’s Disease. In Encyclopedia. https://encyclopedia.pub/entry/45082
Segura-Aguilar, Juan and Bengt Mannervik. "Neurodegeneration in Parkinson’s Disease." Encyclopedia. Web. 31 May, 2023.
Copy Citation
Investigations of the effect of antioxidants on idiopathic Parkinson’s disease have been unsuccessful because the preclinical models used to propose these clinical studies do not accurately represent the neurodegenerative process of the disease. Treatment with certain exogenous neurotoxins induces massive and extremely rapid degeneration; for example, MPTP causes severe Parkinsonism in just three days, while the degenerative process of idiopathic Parkinson´s disease proceeds over many years. The endogenous neurotoxin aminochrome seems to be a good alternative target since it is formed in the nigrostriatal system neurons where the degenerative process occurs.
dopamine
Parkinson’s disease
neuromelanin
antioxidants
1. Mechanisms Involved in Neurodegeneration in Parkinson’s Disease
Although 65 years have passed since the discovery that the motor symptoms of idiopathic Parkinson’s disease are related to the massive loss of neuromelanin-containing neurons in the nigrostriatal system, it is still unclear what triggers the degenerative process of this neuronal system [1][2]. The first widely studied mechanism was the role of oxidative stress in this disease, then mitochondrial dysfunction, and, in the early 1990s, the existence of a mutation in the alpha-synuclein gene was discovered in some families with genetic Parkinson’s. This discovery was a major boost in basic research as it was the first gene with mutations that induced familial or genetic Parkinson’s [3]. Subsequently, more genes related to familial Parkinson’s disease were identified, such as the parkin gene, PINK-1, LRRK2, VPS35, DJ1, ATP13A2, etc. [4]. Although the origin of neurodegeneration in the nigrostriatal system is unclear, there is a general consensus that certain mechanisms are involved in the loss of neuromelanin-containing dopaminergic neurons. They are related to the appearance of motor symptoms and involve oxidative stress, mitochondrial dysfunction, aggregation of alpha-synuclein to neurotoxic oligomers, dysfunction of both lysosomal and proteasomal protein degradation systems, endoplasmic reticulum stress, and neuroinflammation [5][6][7][8].
2. Preclinical Model for Idiopathic Parkinson’s Disease
The preclinical model for idiopathic Parkinson’s disease should use (i) an endogenous neurotoxin that is formed within the neuromelanin-containing dopaminergic neurons of the nigrostriatal system; (ii) a neurotoxin that induces a focal (non-expansive) degeneration; (iii) it should trigger all the mechanisms involved in the degenerative process, such as oxidative stress, mitochondrial dysfunction, alpha-synuclein aggregation to neurotoxic oligomers, protein degradation dysfunction (lysosomal and proteasomal system), endoplasmic reticulum stress, and neuroinflammation. As researchers have seen previously, the only one of the endogenous neurotoxins that researchers have previously described that meets these requirements is aminochrome.
Aminochrome is neurotoxic by being one-electron reduced to leukoaminochrome o-semiquinone radical by most flavoenzymes with the exception of DT-diaphorase (NADP(H):quinone oxidoreductase), which is the unique flavoenzyme that reduces quinones with two electrons [9]. In studies performed with electron spin resonance (ESR), NADPH cytochrome P450 reductase was found to reduce both dopamine ortho-quinone and aminochrome to the free radicals dopamine o-semiquinone and leukoaminochrome o-semiquinone, respectively. The ESR spectrum of dopamine o-semiquinone and of leukoaminochrome o-semiquinone radical was detected at 1 min. However, at 2 min only the dopamine o-semiquinone radical spectrum was detectable by ESR due to the high reactivity of leukoaminochrome o-semiquinone radical with oxygen [10]. Aminochrome one-electron reduction to the leukoaminochrome o-semiquinone radical induces oxidative stress due to the extremely rapid reaction with oxygen, which causes re-oxidation to aminochrome. This generates a redox cycling between aminochrome and leukoaminochrome o-semiquinone radical that functions until cellular dioxygen and/or NAD(P)H are depleted. This redox cycling generates oxidative stress and the depletion of cytosolic NADH for its use to generate ATP in the mitochondrial electron transport chain (Figure 1).
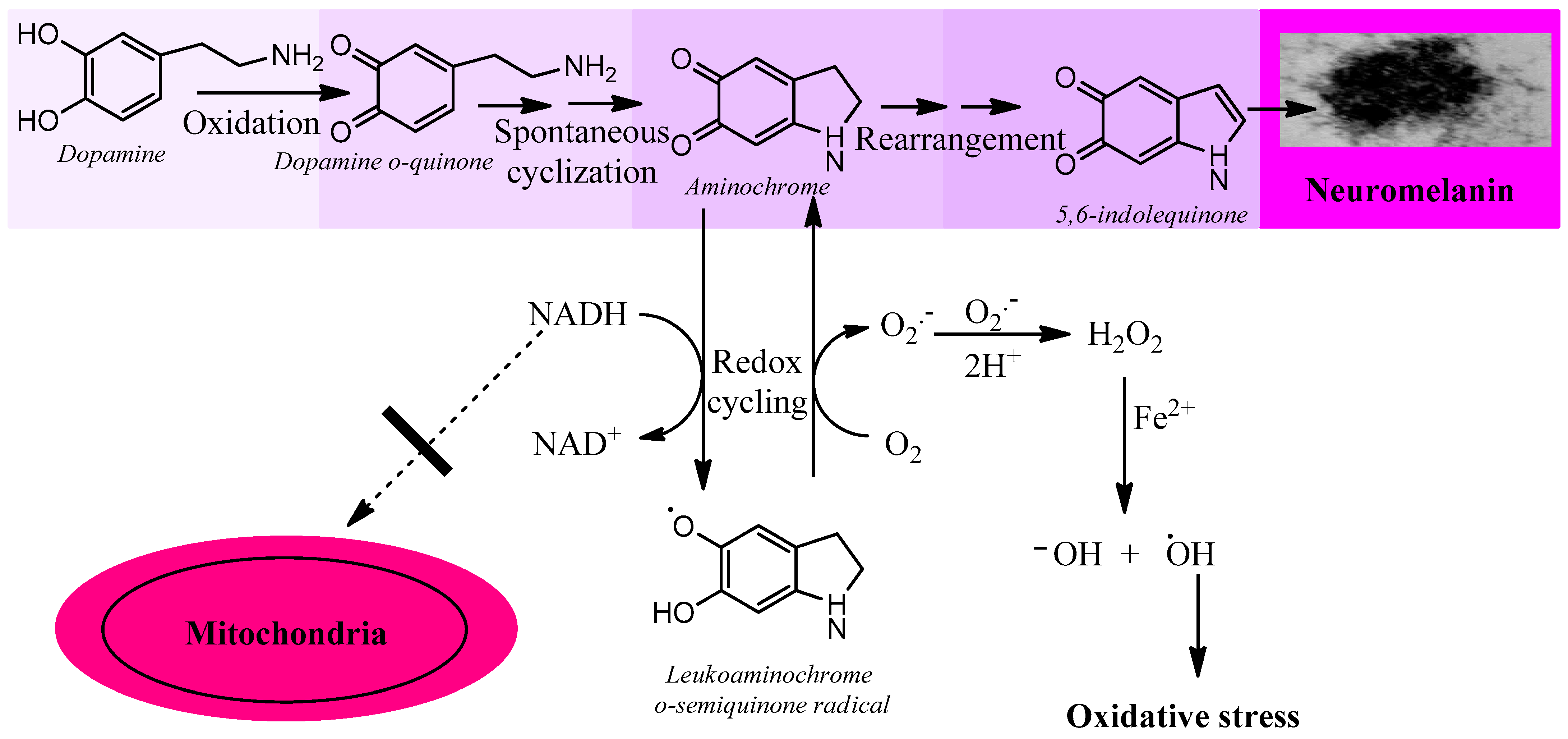
Figure 1. Aminochrome induces oxidative stress. Aminochrome can be reduced with one electron to produce the leukoaminochrome o-semiquinone radical by flavoenzymes using the cytosolic NADH generated to be used in the mitochondria to generate ATP through the transport chain coupled to oxidative phosphorylation from ADP to ATP. The leukoaminochrome o-semiquinone radical is extremely unstable in the presence of dioxygen and autoxidizes to regenerate aminochrome, which is reduced again, creating a redox cycle between aminochrome and leukoaminochrome o-semiquinone radical. This redox cycling is very fast and depletes as much oxygen as NADH with the concomitant production of oxidative stress.
Aminochrome is also neurotoxic by forming covalent complexes with proteins such as alpha-synuclein that induce the formation of neurotoxic oligomers [11]. Aminochrome forms adducts also with actin and α- and β-tubulin disrupting the cytoskeleton architecture [12], which plays an important role in microtubule formation [13]. Aminochrome inhibits complex I of the mitochondrial respiratory chain, resulting in the inhibition of ATP production and mitochondrial dysfunction [14][15]. Aminochrome induces protein degradation dysfunction by inhibiting lysosomal and proteasomal systems [16][17][18]. Aminochrome induces lysosomal dysfunction by inhibiting the vacuolar-type H+-ATPase that plays an essential role in maintaining an acidic pH by pumping protons into lysosomes [19]. Aminochrome also induces endoplasmic reticulum stress and neuroinflammation [16][20][21].
Aminochrome in vivo induces neuronal dysfunction as a consequence of a significant decrease in dopamine release with concomitant enhanced GABA release [22]. Aminochrome induces mitochondrial dysfunction with a significant decrease in ATP production, affecting axonal transport of monoaminergic vesicles for neurotransmission to the terminals that explain the significant decrease in the number of these vesicles in the terminals compared with saline as control. Aminochrome induces a progressive degeneration of dopaminergic neurons while the striatal dopaminergic fibers are intact, accompanied by a dramatic change of tyrosine positive neuron morphology, a phenomenon known as cell shrinkage [22].
The ideal model for idiopathic Parkinson’s disease should consider that the neurotoxin that triggers the degenerative process is generated within the same neuromelanin-containing dopaminergic neuron and induces a degenerative process focused on a single neuron. This last requirement is practically impossible to achieve, since the injection of aminochrome into the striatum or substantia nigra will affect all the neurons that are in contact with aminochrome solution. However, if aminochrome is the neurotoxin that triggers neurodegeneration in idiopathic Parkinson’s, this model could be a good target to search for possible pharmacological compounds that prevent, inhibit, or slow down this degenerative process in idiopathic Parkinson’s.
There is a study in which the human enzyme tyrosinase was overexpressed in the substantia nigra of rats, exacerbating the production of neuromelanin in this region of the brain in all types of neurons and glial cells, inducing nigrostriatal neurodegeneration, hypokinesia, and Lewy body-like formation [23]. The problem with this model is that (i) this overexpression affects the majority of dopaminergic neurons, generating a massive effect on the degenerative process; (ii) the oxidative effect of tyrosinase is not specific for dopaminergic neurons since this overexpression affects all neurons and glial cells. Tyrosinase catalyzes the oxidation of monophenols or diphenols in the presence of dioxygen without the need for a cofactor; however, its catalytic activity with diphenols is higher than with monophenols [24]. Therefore, indiscriminate overexpression in all types of neurons and glial cells reduces the specificity of the model, since tyrosinase can oxidize monophenols in other types of neurons or glial cells with unknown effects. In the case of overexpression of tyrosinase in astrocytes, this enzyme can also oxidize dopamine, since this type of glial cell can take up dopamine released during neurotransmission; (iii) the triggering agent of the neurodegenerative process in this preclinical model is not a neurotoxin that can be the target of new pharmacological drugs attempting to stop the degenerative process, but rather it is tyrosinase, which is not expressed in the human substantia nigra. However, there are some reports that suggest its expression in this tissue [25], but if it were true that tyrosinase is normally expressed in neuromelanin-containing dopaminergic neurons, the presence of the pigment would be observed from an early age in children. Several studies have indeed shown that tyrosinase is not present in the human substantia nigra, even using highly specific and sensitive mass spectroscopy [26][27] (Zucca et al., 2018; Tribl et al., 2007). It has been proposed that animals that produce more neuromelanin and more closely resemble the human brain such as sheep and goats are more appropriate as a preclinical animal model for Parkinson’s disease [28] (Capucciati et al., 2021).
3. Transcriptional Gene Activation via KEAP1/NRF2
Transcriptional activation of genes encoding a battery of cellular defense enzymes that provide protection against oxidative and electrophilic stress is affected by the transcription factor NRF2 (nuclear factor (erythroid-derived 2)-like 2) binding to an antioxidant responsive element or an electrophile responsive element (ARE/EpRE). Among the various cytoprotective proteins are γ-glutamylcysteine ligase, GSTs, and DT-diaphorase [29][30]. NFR2 is bound to KEAP1 (Kelch-like ECH-associated protein 1) in the cytosol, where it is directed to rapid proteasomal degradation. KEAP1 senses reactions of its sulfhydryl groups with electrophiles and oxidants and, as a consequence, NRF2 is released and targets ARE/ErRE elements of DNA in the nucleus. KEAP1/NRF2 is recognized as the most prominent protective system against electrophilic and oxidative insults; the upregulation of key enzymes in the defense against toxicants implicated in Parkinson’s disease and other neurodegenerative conditions is noteworthy. For obvious reasons, the KEAP1/NFR2 system has been considered for pharmacological interventions in Parkinson’s disease [31][32], Figure 2.
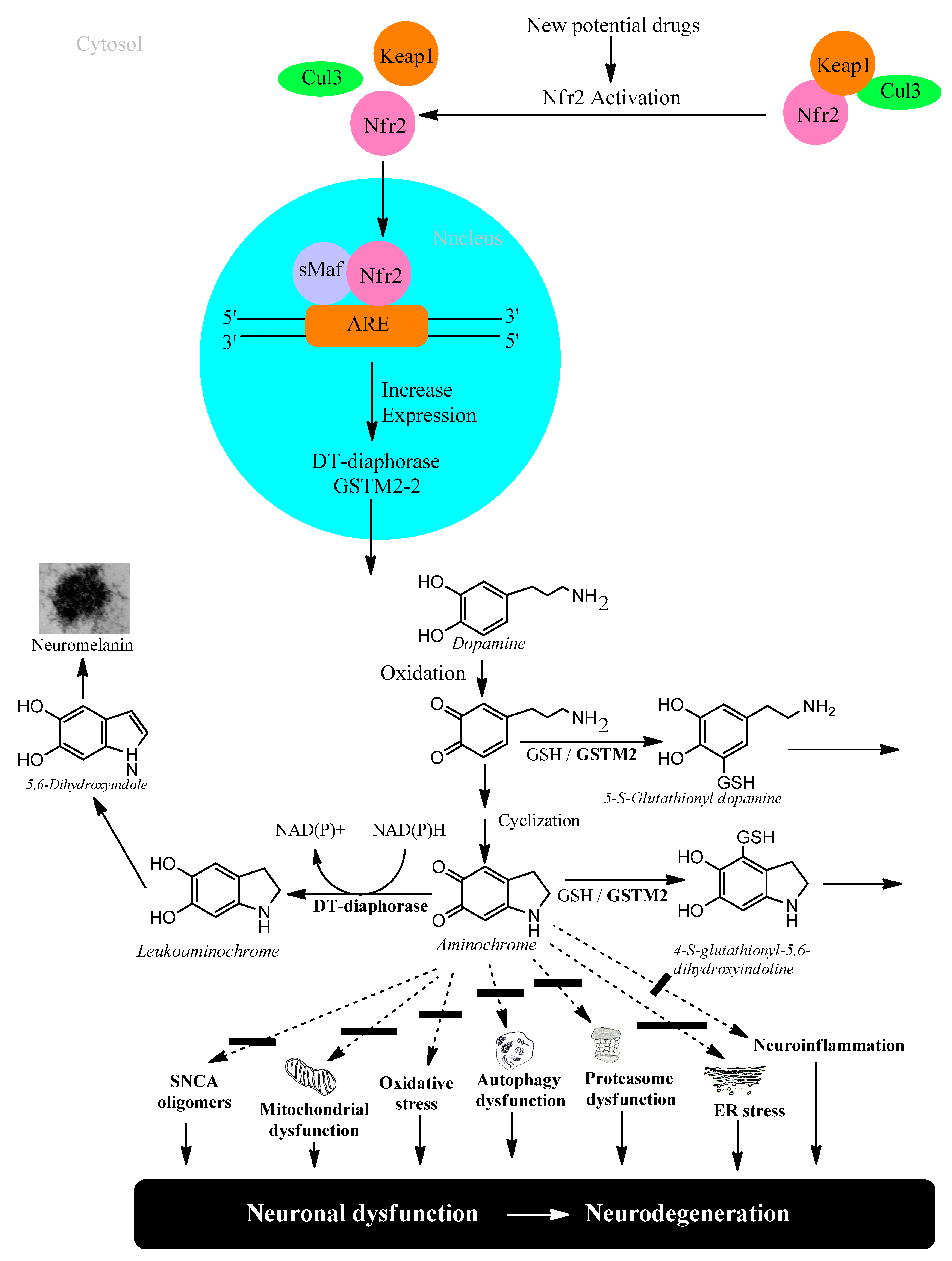
Figure 2. NFR2 activation to protect dopaminergic neurons from aminochrome neurotoxicity.
4. Neuroprotection against Neurodegeneration of Nigrostriatal System in Parkinson’s Disease
Studies carried out with postmortem material from healthy elderly people show intact neuromelanin-containing dopaminergic neurons, suggesting that neuromelanin synthesis is a natural and harmless process. Neuromelanin has been observed to increase over the years in the human brain [33]. However, there is an apparent contradiction, since the synthesis of neuromelanin requires the oxidation of dopamine to three ortho-quinones that can be neurotoxic. Among these aminochrome is the most toxic that induces oxidative stress, the formation of neurotoxic alpha-synuclein oligomers, dysfunction of both lysosomal and proteasomal protein breakdown systems, plasma reticulum stress, mitochondrial dysfunction, and neuroinflammation. The reason why the elderly have their neuromelanin-containing dopaminergic neurons intact at the time of death is because there are two enzymes that prevent the neurotoxic effects of aminochrome DT-diaphorase and glutathione transferase M2-2.
4.1. DT-Diaphorase
DT-diaphorase (NAD(P)H: quinone oxidoreductase, NQO1) is a flavoenzyme with FAD as a prosthetic group that transfers two electrons from either NADH or NADPH to quinones, thereby reducing them to hydroquinones [9][26][34]. This enzyme is 90% located in the cytosol and 5% associated with the endoplasmic reticulum and mitochondria. It is widely expressed in different organs; in the brain it is expressed in the substantia nigra, striatum, cortex, hypothalamus, and hippocampus. In rat substantia nigra, DT-diaphorase is responsible for 97% of the total quinone reductase activity. DT-diaphorase is expressed in tyrosine hydroxylase positive neurons and in astrocytes. DT-diaphorase reduces aminochrome to leukoaminochrome, preventing its neurotoxic effects in a catecholaminergic cell line [35], but DT-diaphorase also prevents aminochrome induced-toxicity in a human astrocyte cell line [36]. DT-diaphorase prevents aminochrome-induced oxidative stress, mitochondrial dysfunction, formation of neurotoxic alpha-synuclein oligomers, proteasome dysfunction, autophagy dysfunction, and lysosome dysfunction [11][12][16][17][19][21][27][30][37][38][39][40].
4.2. Glutathione Transferase M2-2
Electrophilic compounds such as epoxides, alkenals, and quinones can react with GSH and thereby become inactivated and form glutathione conjugates suitable for export from the cytosol [41][42]. The reactions are catalyzed by 20-odd enzymes, which are differentially distributed in cells and tissues. In humans and other mammals, the GSTs have been grouped into membrane-bound (microsomal) and soluble (cytosolic) proteins [43][44]. The former are members of the MAPEG (membrane-associated proteins in eicosanoid and glutathione metabolism) family [45][46]. The soluble GSTs are dimeric proteins occurring in eight classes of homologous sequences, which are catalytically active as homodimers as well as heterodimers [47][48]. The dimers are thus denoted according to their subunit composition, for example, as GST M1-1, GST M1-2, and GST M2-2 encoded by the GSTM1 and GSTM2 genes of the Mu class. With respect to the large variety of compounds identified as substrates for the various GSTs, it is noteworthy that only GST M2-2 has been found to catalyze with high efficiency the conjugation of ortho-quinones derived from dopamine [49][50][51]. The most active glutathione transferase catalyzing aminochrome conjugation is glutathione transferase M2-2, which gives rise to 4-S-glutathionyl-5,6-dihydroxyindoline. Interestingly, 4-S-glutathionyl-5,6-dihydroxyindoline is not oxidized by physiologically occurring oxidants such as dioxygen, hydrogen peroxide, and superoxide. Glutathione transferase M2-2 also catalyzes glutathione conjugation of the aminochrome precursor dopamine ortho-quinone to 5-glutathionyl dopamine that is degraded to 5-cysteinyl dopamine, which has been detected in human cerebrospinal fluid and neuromelanin [46][48][49][50][51][52][53][54][55] (Figure 3).
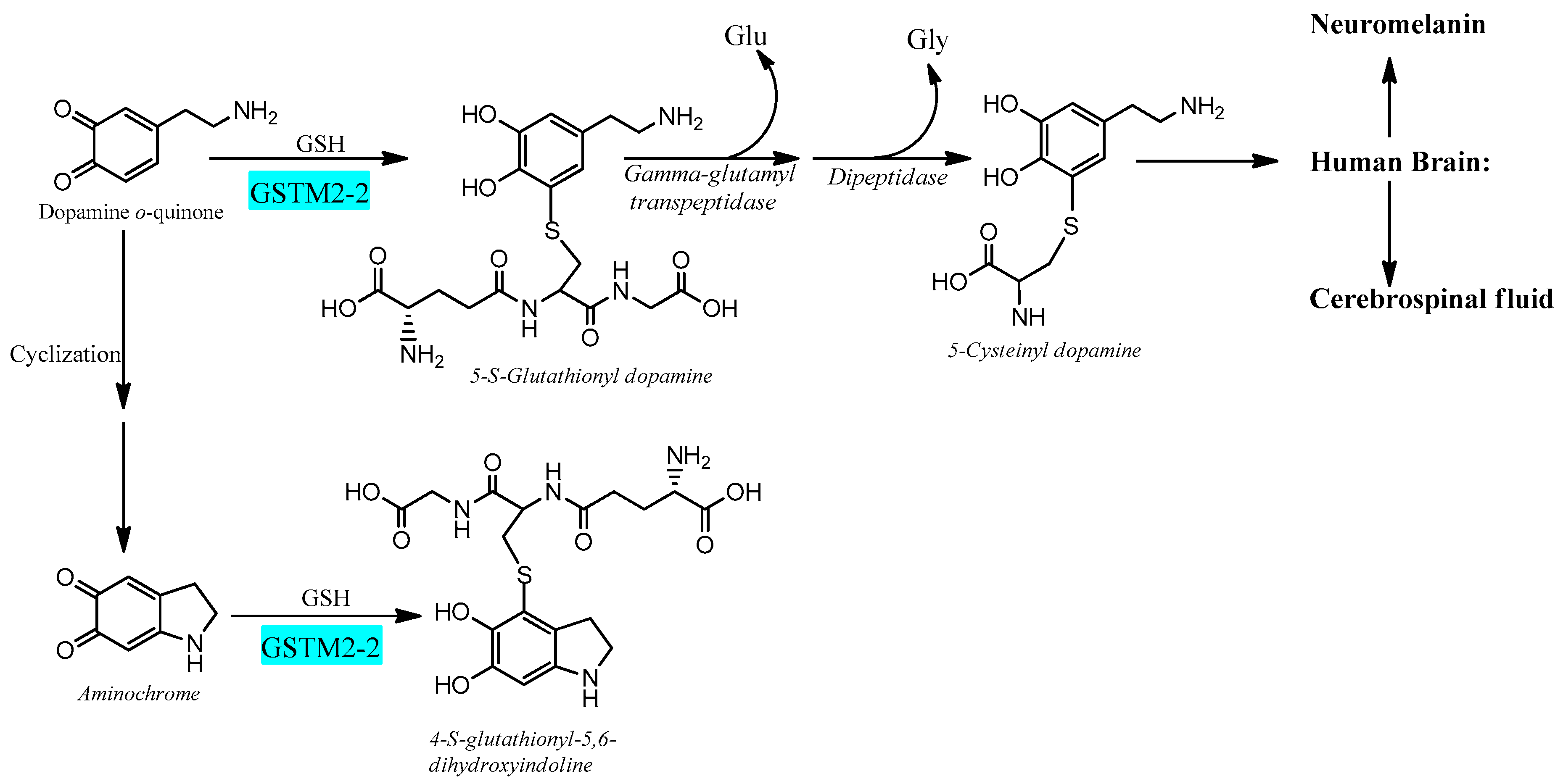
Figure 3. Glutathione transferase M2-2 catalyzes glutathione conjugation of ortho-quinones.
However, whether 5-cysteinyl is an end product is controversial as it has been reported by in vitro experiments that 5-cysteinyldopamine may be neurotoxic by being oxidized to 5-cysteinyldopamine o-quinone, which is converted to a bicyclic o-quinone imine [53][56]. If this mechanism is correct, it remains to explain the presence of 5-cysteinyldopamine in human cerebrospinal fluid and neuromelanin [54][55], since 5-cysteinyldopamine is converted to bicyclic o-quinone imine [56].
4.3. Astrocytes Protect Dopaminergic Neurons against Aminochrome Neurotoxicity
The brain is responsible for more than 20% of the energy consumption in the human body. The organism derives ATP via oxidation of carbohydrates, fats, and proteins and by coupling electron transport with phosphorylation of ADP to ATP. Neurons require ATP to transport proteins and neurotransmission vesicles to neuronal terminals, and neurotransmission itself is completely dependent on ATP. In other words, neuronal function is absolutely dependent on the presence of ATP, which, to a large extent, is generated in the mitochondria. Electrons in the transport chain from NADH flowing through complex I or complex II are further transmitted to ubiquinone Q10 (also known as coenzyme Q10), which in turn transfers the electrons to complex III. In this transfer of electrons, ubiquinone Q10 is reduced with one electron to the free radical ubiquinone Q10 semiquinone. The latter is subsequently reduced to ubiquinone Q10 hydroquinone, which transfers the electrons to complex III. However, the radical ubiquinone Q10 semiquinone is able to reduce dioxygen to superoxide, generating electron transport leakage and oxidative stress. Superoxide enzymatically or spontaneously can generate hydrogen peroxide, which, in the presence of reduced iron, forms hydroxyl radicals. This leakage of electrons from the mitochondrial electron transport chain permanently exposes neurons to conditions of oxidative stress. Astrocytes play an important role in protecting neurons against oxidative stress since astrocytes secrete precursors of glutathione, which is an important antioxidant [54][57][58]. Neuromelanin-containing dopaminergic neurons are also exposed to the neurotoxic effects of aminochrome, leading to oxidative stress, mitochondrial dysfunction, formation of neurotoxic alpha-synuclein oligomers, dysfunction of both lysosomal and proteasomal protein degradation systems, neuroinflammation, and endoplasmic reticulum stress [14][15][16][17][20][21][27][28]. However, astrocytes also play a neuroprotective role for neuromelanin-containing dopaminergic neurons by secreting exosomes loaded with the enzyme glutathione transferase M2-2 that penetrate dopaminergic neurons to, in concert with DT-diaphorase, prevent the neurotoxic effects of aminochrome [48][54][55][57][58][59] (Figure 4).
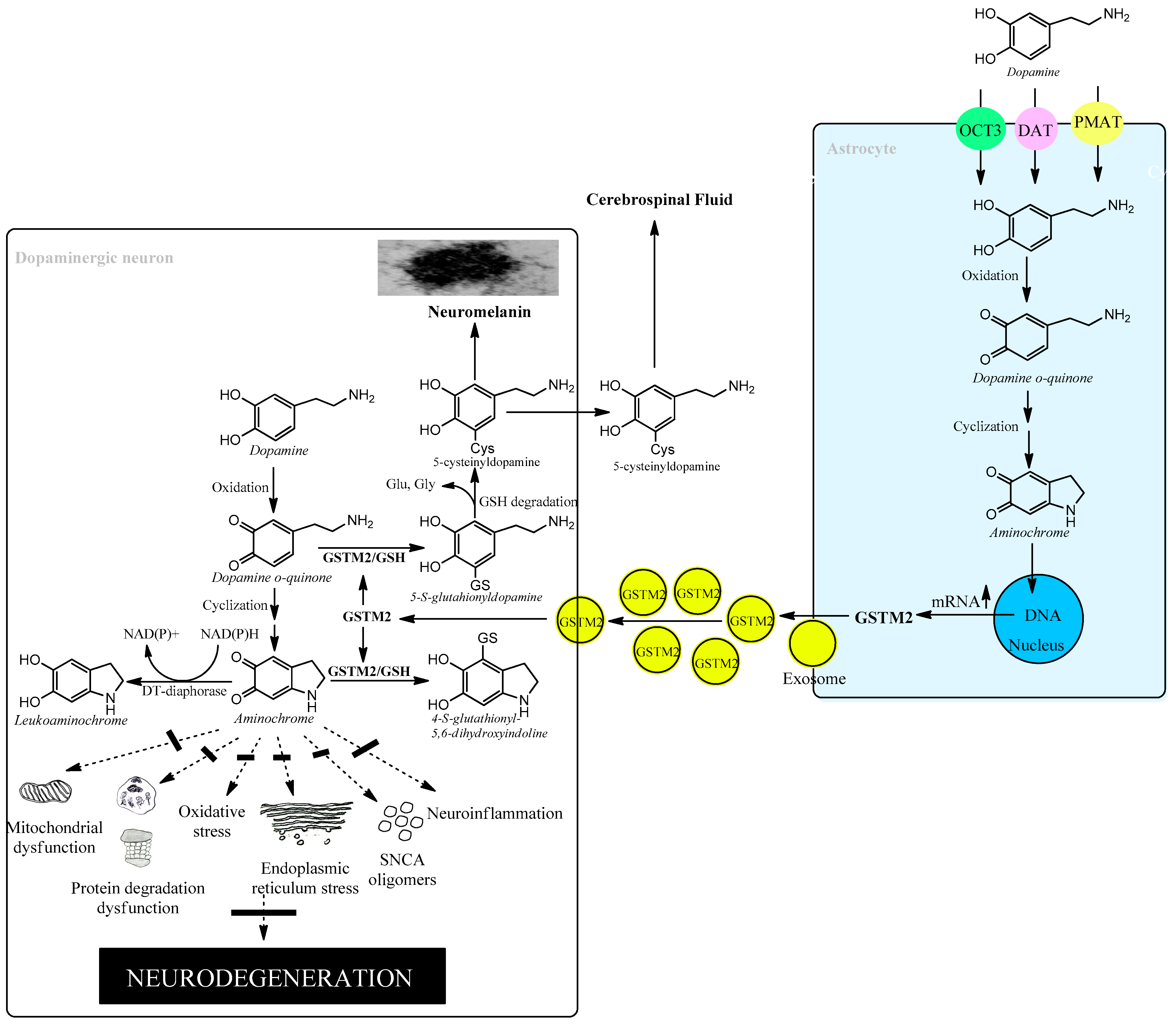
Figure 4. Astrocytes protect dopaminergic neurons against aminochrome neurotoxicity.
4.4. Glutathione and Oxidative Stress
Glutathione (GSH) is a fundamental endogenous constituent of the molecular defense that provides protection against electrophilic agents and oxidative stress [1][60]. GSH, L-γ-glutamyl-L-cysteinylglycine, is formed by the sequential actions of γ-glutamylcysteine ligase and glutathione synthetase, of which reactions the first is rate-limiting in GSH biosynthesis [2][61]. It has been demonstrated that GSH is ubiquitously present in the brain and that substantia nigra in Parkinson’s patients is selectively depleted in GSH content compared with healthy individuals [3][4][62][63]. Whether the loss of GSH is a cause or an effect of the disease remains unknown but attempts to raise the GSH concentration by pharmacological intervention have been made under the premise that an elevated GSH level would provide a therapeutic consequence [5][64]. In general, GSH cannot cross cell membranes or penetrate the blood–brain barrier and oral administration has been ruled out. Nasal spray delivery of GSH has been attempted, but the efficacy is unclear [6][65]. The amino acid limiting the biosynthesis of GSH is cysteine, which is normally transported into cells as the disulfide cystine via the system xc-cystine/glutamate antiporter [7][66]. Thus, intravenous administration of the precursor N-acetylcysteine has been reported to increase the GSH concentration in the brain [8][67]. The characteristic pathological features of substantia nigra in Parkinson’s patients comprise enhanced iron deposition and lipid peroxidation, as wells as defects in the transport system xc-. These attributes are congruent with the hallmarks of ferroptosis, which is currently receiving attention in various diseases [68][69]. Preventing the progressive loss of dopaminergic neurons by interfering with ferroptosis is a new approach to treatment [6][10][65][70]. GSH can serve as an antioxidant and five selenium-dependent peroxidases (GPXs) catalyze the reduction of hydrogen peroxide and various lipid hydroperoxides [37][71]. The enzymes play a prominent role in the protection against reactive oxygen species (ROS). GPX4 is particularly active with phospholipid hydroperoxides and appears to play an essential function in preventing ferroptosis [68][69].
References
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42.
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology; Pathology, Genetics, a50nd Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12.
- Yoo, J.M.; Lin, Y.; Heo, Y.; Lee, Y.H. Polymorphism in alpha-synuclein oligomers and its implications in toxicity under disease conditions. Front. Mol. Biosci. 2022, 9, 959425.
- Day, J.O.; Mullin, S. The Genetics of Parkinson’s Disease and Implications for Clinical Practice. Genes 2021, 12, 1006.
- Mercado, G.; Castillo, V.; Soto, P.; Sidhu, A. ER stress and Parkinson disease: Pathological inputs that converge into the secretory pathway. Brain Res. 2016, 16, 30260–30268.
- Moors, T.; Paciotti, S.; Chiasserini, D.; Calabresi, P.; Parnetti, L.; Beccari, T.; van de Berg, W.D. Lysosomal Dysfunction and α-Synuclein Aggregation in Parkinson’s Disease. Diagnostic. Links Mov. Disord. 2016, 6, 791–801.
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109 Pt B, 249–257.
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808.
- Segura-Aguilar, J. Neuroprotective mechanisms against dopamine oxidation-dependent neurotoxicity. In Clinical Studies and Therapies in Parkinson’s Disease: Translations from Preclinical Models; Segura-Aguilar, J., Ed.; Elsevier: Cambridge, MA, USA, 2021; pp. 229–240.
- Segura-Aguilar, J.; Metodiewa, D.; Welch, C.J. Metabolic activation of dopamine o-quinones to o-semiquinones by NADPH cytochrome P450 reductase may play an important role in oxidative stress and apoptotic effects. Biochim. Biophys. Acta 1998, 1381, 1–6.
- Muñoz, P.; Cardenas, S.; Huenchuguala, S.; Briceño, A.; Couve, E.; Paris, I.; Segura-Aguilar, J. DT-Diaphorase Prevents Aminochrome-Induced Alpha-Synuclein Oligomer Formation and Neurotoxicity. Toxicol. Sci. 2015, 145, 37–47.
- Paris, I.; Perez-Pastene, C.; Cardenas, S.; Iturriaga-Vasquez, P.; Muñoz, P.; Couve, E.; Caviedes, P.; Segura-Aguilar, J. Aminochrome induces disruption of actin, alpha-, and beta-tubulin cytoskeleton networks in substantia-nigra-derived cell line. Neurotox. Res. 2010, 18, 82–92.
- Briceño, A.; Muñoz, P.; Brito, P.; Huenchuguala, S.; Segura-Aguilar, J.; Paris, I.B. Aminochrome Toxicity is Mediated by Inhibition of Microtubules Polymerization Through the Formation of Adducts with Tubulin. Neurotox. Res. 2016, 29, 381–393.
- Aguirre, P.; Urrutia, P.; Tapia, V.; Villa, M.; Paris, I.; Segura-Aguilar, J.; Núñez, M.T. The dopamine metabolite aminochrome inhibits mitochondrial complex I and modifies the expression of iron transporters DMT1 and FPN1. Biometals 2012, 25, 795–803.
- Paris, I.; Muñoz, P.; Huenchuguala, S.; Couve, E.; Sanders, L.H.; Greenamyre, J.T.; Caviedes, P.; Segura-Aguilar, J. Autophagy protects against aminochrome-induced cell death in substantia nigra-derived cell line. Toxicol. Sci. 2011, 121, 376–388.
- Zafar, K.S.; Inayat-Hussain, S.H.; Siegel, D.; Bao, A.; Shieh, B.; Ross, D. Overexpression of NQO1 protects human SK-N-MC neuroblastoma cells against dopamine-induced cell death. Toxicol. Lett. 2006, 166, 261–267.
- Huenchuguala, S.; Muñoz, P.; Zavala, P.; Villa, M.; Cuevas, C.; Ahumada, U.; Graumann, R.; Nore, B.; Couve, E.; Mannervik, B.; et al. Glutathione transferase mu 2 protects glioblastoma cells against aminochrome toxicity by preventing autophagy and lysosome dysfunction. Autophagy 2014, 10, 618–630.
- Muñoz, P.; Huenchuguala, S.; Paris, I.; Segura-Aguilar, J. Dopamine oxidation and autophagy. Parkinsons. Dis. 2012, 2012, 920953.
- Meléndez, C.; Muñoz, P.; Segura-Aguilar, J. DT-Diaphorase Prevents Aminochrome-Induced Lysosome Dysfunction in SH-SY5Y Cells. Neurotox. Res. 2019, 35, 255–259.
- Santos, C.C.; Araújo, F.M.; Ferreira, R.S.; Silva, V.B.; Silva, J.H.C.; Grangeiro, M.S.; Soares, É.N.; Pereira, É.P.L.; Souza, C.S.; Costa, S.L.; et al. Aminochrome induces microglia and astrocyte activation. Toxicol. In Vitro 2017, 42, 54–60.
- Xiong, R.; Siegel, D.; Ross, D. Quinone-induced protein handling changes: Implications for major protein handling systems in quinone-mediated toxicity. Toxicol. Appl. Pharmacol. 2014, 280, 285–295.
- Herrera, A.; Muñoz, P.; Paris, I.; Díaz-Veliz, G.; Mora, S.; Inzunza, J.; Hultenby, K.; Cardenas, C.; Jaña, F.; Raisman-Vozari, R.; et al. Aminochrome induces dopaminergic neuronal dysfunction: A new animal model for Parkinson’s disease. Cell. Mol. Life Sci. 2016, 73, 3583–3597.
- Carballo-Carbajal, I.; Laguna, A.; Romero-Giménez, J.; Cuadros, T.; Bové, J.; Martinez-Vicente, M.; Parent, A.; Gonzalez-Sepulveda, M.; Peñuelas, N.; Torra, A.; et al. Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson’s disease pathogenesis. Nat. Commun. 2019, 10, 973.
- Do, H.; Kang, E.; Yang, B.; Cha, H.J.; Choi, Y.S. A tyrosinase, mTyr-CNK, that is functionally available as a monophenol monooxygenase. Sci. Rep. 2017, 7, 17267.
- Yamamuro, Y.; Ogura, S. Regional expression of tyrosinase in central catecholaminergic systems of colored mice. Exp. Anim. 2019, 68, 49–56.
- Tribl, F.; Arzberger, T.; Riederer, P.; Gerlach, M. Tyrosinase is not detected in human catecholaminergic neurons by immunohistochemistry and Western blot analysis. J. Neural. Transm. Suppl. 2007, 72, 51–55.
- Zucca, F.A.; Vanna, R.; Cupaioli, F.A.; Bellei, C.; De Palma, A.; Di Silvestre, D.; Mauri, P.; Grassi, S.; Prinetti, A.; Casella, L.; et al. Neuromelanin organelles are specialized autolysosomes that accumulate undegraded proteins and lipids in aging human brain and are likely involved in Parkinson’s disease. NPJ Parkinsons. Dis. 2018, 4, 7.
- Capucciati, A.; Zucca, F.A.; Monzani, E.; Zecca, L.; Casella, L.; Hofer, T. Interaction of Neuromelanin with Xenobiotics and Consequences for Neurodegeneration; Promising Experimental Models. Antioxidants 2021, 10, 824.
- Ghosh, A.; Chandran, K.; Kalivendi, S.V.; Joseph, J.; Antholine, W.E.; Hillard, C.J.; Kanthasamy, A.; Kanthasamy, A.; Kalyanaraman, B. Neuroprotection by a mitochondria-targeted drug in a Parkinson’s disease model. Free Radic. Biol. Med. 2010, 49, 1674–1684.
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203.
- Xi, Y.; Feng, D.; Tao, K.; Wang, R.; Shi, Y.; Qin, H.; Murphy, M.P.; Yang, Q.; Zhao, G. MitoQ protects dopaminergic neurons in a 6-OHDA induced PD model by enhancing Mfn2-dependent mitochondrial fusion via activation of PGC-1α. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2859–2870.
- Yang, X.X.; Yang, R.; Zhang, F. Role of Nrf2 in Parkinson’s Disease: Toward New Perspectives. Front. Pharmacol. 2022, 13, 919233.
- Zecca, L.; Fariello, R.; Riederer, P.; Sulzer, D.; Gatti, A.; Tampellini, D. The absolute concentration of nigral neuromelanin, assayed by a new sensitive method, increases throughout the life and is dramatically decreased in Parkinson’s disease. FEBS Lett. 2002, 510, 216–220.
- Segura-Aguilar, J.; Muñoz, P.; Inzunza, J.; Varshney, M.; Nalvarte, I.; Mannervik, B. Neuroprotection against Aminochrome Neurotoxicity: Glutathione Transferase M2-2 and DT-Diaphorase. Antioxidants 2022, 11, 296.
- Lozano, J.; Muñoz, P.; Nore, B.F.; Ledoux, S.; Segura-Aguilar, J. Stable expression of short interfering RNA for DT-diaphorase induces neurotoxicity. Chem. Res. Toxicol. 2010, 23, 1492–1496.
- Huenchuguala, S.; Muñoz, P.; Graumann, R.; Paris, I.; Segura-Aguilar, J. DT-diaphorase protects astrocytes from aminochrome-induced toxicity. Neurotoxicology 2016, 55, 10–12.
- Arriagada, C.; Paris, I.; Sanchez de las Matas, M.J.; Martinez-Alvarado, P.; Cardenas, S.; Castañeda, P.; Graumann, R.; Perez-Pastene, C.; Olea-Azar, C.; Couve, E.; et al. On the neurotoxicity mechanism of leukoaminochrome o-semiquinone radical derived from dopamine oxidation: Mitochondria damage, necrosis, and hydroxyl radical formation. Neurobiol. Dis. 2004, 16, 468–477.
- Kostrzewa, R.M.; Jacobowitz, D.M. Pharmacological actions of 6-hydroxydopamine. Pharmacol. Rev. 1974, 26, 199–288.
- Biesemeier, A.; Eibl, O.; Eswara, S.; Audinot, J.N.; Wirtz, T.; Pezzoli, G.; Zucca, F.A.; Zecca, L.; Schraermeyer, U. Elemental mapping of Neuromelanin organelles of human Substantia Nigra: Correlative ultrastructural and chemical analysis by analytical transmission electron microscopy and nano-secondary ion mass spectrometry. J. Neurochem. 2016, 138, 339–353.
- Segura-Aguilar, J. Dopamine oxidation to neuromelanin and neurotoxic metabolites. In Clinical Studies and Therapies in Parkinson’s Disease: Translations from Preclinical Models; Segura-Aguilar, J., Ed.; Elsevier: Cambridge, MA, USA, 2021; pp. 213–227.
- Musatov, A.; Robinson, N.C. Susceptibility of mitochondrial electron-transport complexes to oxidative damage. Focus on cytochrome c oxidase. Free Radic. Res. 2012, 46, 1313–1326.
- Mannervik, B. Evolution of glutathione transferases and related enzymes for the protection of cells against electrophiles. Biochem. Soc. Trans. 1996, 24, 878–880.
- Jing, L.; He, M.T.; Chang, Y.; Mehta, S.L.; He, Q.P.; Zhang, J.Z.; Li, P.A. Coenzyme Q10 protects astrocytes from ROS-induced damage through inhibition of mitochondria-mediated cell death pathway. Int. J. Biol. Sci. 2015, 11, 59–66.
- Mannervik, B.; Board, P.G.; Hayes, J.D.; Listowsky, I.; Pearson, W.R. Nomenclature for mammalian soluble glutathione transferases. Methods Enzymol. 2005, 401, 1–8.
- Beal, M.F. Coenzyme Q10 administration and its potential for treatment of neurodegenerative diseases. Biofactors 1999, 9, 261–266.
- Jakobsson, P.J.; Morgenstern, R.; Mancini, J.; Ford-Hutchinson, A.; Persson, B. Common structural features of MAPEG—A widespread superfamily of membrane associated proteins with highly divergent functions in eicosanoid and glutathione metabolism. Protein Sci. 1999, 8, 689–692.
- Park, H.W.; Park, C.G.; Park, M.; Lee, S.H.; Park, H.R.; Lim, J.; Paek, S.H.; Choy, Y.B. Intrastriatal administration of coenzyme Q10 enhances neuroprotection in a Parkinson’s disease rat model. Sci. Rep. 2020, 10, 9572.
- Mannervik, B.; Jensson, H. Binary combinations of four protein subunits with different catalytic specificities explain the relationship between six basic glutathione S-transferases in rat liver cytosol. J. Biol. Chem. 1982, 257, 9909–9912.
- Yoritaka, A.; Kawajiri, S.; Yamamoto, Y.; Nakahara, T.; Ando, M.; Hashimoto, K.; Nagase, M.; Saito, Y.; Hattori, N. Randomized, double-blind, placebo-controlled pilot trial of reduced coenzyme Q10 for Parkinson’s disease. Parkinsonism. Relat. Disord. 2015, 21, 911–916.
- Segura-Aguilar, J.; Baez, S.; Widersten, M.; Welch, C.J.; Mannervik, B. Human class Mu glutathione transferases, in particular isoenzyme M2-2, catalyze detoxication of the dopamine metabolite aminochrome. J. Biol. Chem. 1997, 272, 5727–5731.
- Baez, S.; Segura-Aguilar, J.; Widersten, M.; Johansson, A.S.; Mannervik, B. Glutathione transferases catalyse the detoxication of oxidized metabolites (o-quinones) of catecholamines and may serve as an antioxidant system preventing degenerative cellular processes. Biochem. J. 1997, 324, 25–28.
- Beal, M.F.; Oakes, D.; Shoulson, I.; Henchcliffe, C.; Galpern, W.R.; Haas, R.; Juncos, J.L.; John, G.N.; Tiffini Smith, V.; Bernard, R.; et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol. 2014, 71, 543–552.
- Dagnino-Subiabre, A.; Cassels, B.K.; Baez, S.; Johansson, A.S.; Mannervik, B.; Segura-Aguilar, J. Glutathione transferase M2-2 catalyzes conjugation of dopamine and dopa o-quinones. Biochem. Biophys. Res. Commun. 2000, 274, 32–36.
- Cheng, F.C.; Kuo, J.S.; Chia, L.G.; Dryhurst, G. Elevated 5-S-cysteinyldopamine/homovanillic acid ratio and reduced homovanillic acid in cerebrospinal fluid: Possible markers for and potential insights into the pathoetiology of Parkinson’s disease. J. Neural. Transm. 1996, 103, 433–446.
- Rosengren, E.; Linder-Eliasson, E.; Carlsson, A. Detection of 5-S-cysteinyldopamine in human brain. J. Neural. Transm. 1985, 63, 247–253.
- Mosca, L.; Lendaro, E.; d’Erme, M.; Marcellini, S.; Moretti, S.; Rosei, M.A. 5-S-Cysteinyl-dopamine effect on the human dopaminergic neuroblastoma cell line SH-SY5Y. Neurochem. Int. 2006, 49, 262–269.
- Cuevas, C.; Huenchuguala, S.; Muñoz, P.; Villa, M.; Paris, I.; Mannervik, B.; Segura-Aguilar, J. Glutathione transferase-M2-2 secreted from glioblastoma cell protects SH-SY5Y cells from aminochrome neurotoxicity. Neurotox. Res. 2015, 27, 217–228.
- Valdes, R.; Armijo, A.; Muñoz, P.; Hultenby, K.; Hagg, A.; Inzunza, J.; Nalvarte, I.; Varshney, M.; Mannervik, B.; Segura-Aguilar, J. Cellular Trafficking of Glutathione Transferase M2-2 Between U373MG and SHSY-S7 Cells is Mediated by Exosomes. Neurotox Res. 2021, 39, 182–190.
- Segura-Aguilar, J.; Mannervik, B.; Inzunza, J.; Varshney, M.; Nalvarte, I.; Muñoz, P. Astrocytes protect dopaminergic neurons against aminochrome neurotoxicity. Neural. Regen. Res. 2022, 17, 1861–1866.
- Labarrere, C.A.; Kassab, G.S. Glutathione: A Samsonian life-sustaining small molecule that protects against oxidative stress, ageing and damaging inflammation. Front. Nutr. 2022, 9, 1007816.
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760.
- Jenner, P. Oxidative damage in neurodegenerative disease. Lancet 1994, 344, 796–798.
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355.
- Aoyama, K. Glutathione in the Brain. Int. J. Mol. Sci. 2021, 22, 5010.
- Lin, K.J.; Chen, S.D.; Lin, K.L.; Liou, C.-W.; Lan, M.-Y.; Chuang, Y.-C.; Wang, P.-W.; Lee, J.-J.; Wang, F.-S.; Lin, H.-Y.; et al. Iron Brain Menace: The Involvement of Ferroptosis in Parkinson Disease. Cells 2022, 11, 3829.
- Bridges, R.J.; Natale, N.R.; Patel, S.A. System xc⁻ cystine/glutamate antiporter: An update on molecular pharmacology and roles within the CNS. Br. J. Pharmacol. 2012, 165, 20–34.
- Holmay, M.J.; Terpstra, M.; Coles, L.D.; Mishra, U.; Ahlskog, M.; Öz, G.; Cloyd, J.C.; Tuite, P.J. N-Acetylcysteine boosts brain and blood glutathione in Gaucher and Parkinson diseases. Clin. Neuropharmacol. 2013, 36, 103–106.
- Segura-Aguilar, J. Parkinson´s disease. In Clinical Studies and Therapies in Parkinson’s Disease: Translations from Preclinical Models; Segura-Aguilar, J., Ed.; Elsevier: Cambridge, MA, USA, 2021; pp. 1–175.
- Stockwell, B.R.; Jiang, X.; Gu, W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell. Biol. 2020, 30, 478–490.
- Wang, Z.L.; Yuan, L.; Li, W.; Li, J.Y. Ferroptosis in Parkinson’s disease: Glia-neuron crosstalk. Trends Mol. Med. 2022, 28, 258–269.
- Brigelius-Flohé, R.; Flohé, L. Regulatory Phenomena in the Glutathione Peroxidase Superfamily. Antioxid. Redox. Signal 2020, 33, 498–516.
More
Information
Subjects:
Others
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
01 Jun 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No