Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Juan Segura-Aguilar and Version 2 by Rita Xu.
Investigations of the effect of antioxidants on idiopathic Parkinson’s disease have been unsuccessful because the preclinical models used to propose these clinical studies do not accurately represent the neurodegenerative process of the disease. Treatment with certain exogenous neurotoxins induces massive and extremely rapid degeneration; for example, MPTP causes severe Parkinsonism in just three days, while the degenerative process of idiopathic Parkinson´s disease proceeds over many years. The endogenous neurotoxin aminochrome seems to be a good alternative target since it is formed in the nigrostriatal system neurons where the degenerative process occurs.
- dopamine
- Parkinson’s disease
- neuromelanin
- antioxidants
1. Mechanisms Involved in Neurodegeneration in Parkinson’s Disease
Although 65 years have passed since the discovery that the motor symptoms of idiopathic Parkinson’s disease are related to the massive loss of neuromelanin-containing neurons in the nigrostriatal system, it is still unclear what triggers the degenerative process of this neuronal system [1][2][1,2]. The first widely studied mechanism was the role of oxidative stress in this disease, then mitochondrial dysfunction, and, in the early 1990s, the existence of a mutation in the alpha-synuclein gene was discovered in some families with genetic Parkinson’s. This discovery was a major boost in basic research as it was the first gene with mutations that induced familial or genetic Parkinson’s [3]. Subsequently, more genes related to familial Parkinson’s disease were identified, such as the parkin gene, PINK-1, LRRK2, VPS35, DJ1, ATP13A2, etc. [4]. Although the origin of neurodegeneration in the nigrostriatal system is unclear, there is a general consensus that certain mechanisms are involved in the loss of neuromelanin-containing dopaminergic neurons. They are related to the appearance of motor symptoms and involve oxidative stress, mitochondrial dysfunction, aggregation of alpha-synuclein to neurotoxic oligomers, dysfunction of both lysosomal and proteasomal protein degradation systems, endoplasmic reticulum stress, and neuroinflammation [5][6][7][8][5,6,7,8].
2. Preclinical Model for Idiopathic Parkinson’s Disease
The preclinical model for idiopathic Parkinson’s disease should use (i) an endogenous neurotoxin that is formed within the neuromelanin-containing dopaminergic neurons of the nigrostriatal system; (ii) a neurotoxin that induces a focal (non-expansive) degeneration; (iii) it should trigger all the mechanisms involved in the degenerative process, such as oxidative stress, mitochondrial dysfunction, alpha-synuclein aggregation to neurotoxic oligomers, protein degradation dysfunction (lysosomal and proteasomal system), endoplasmic reticulum stress, and neuroinflammation. As rwesearchers have seen previously, the only one of the endogenous neurotoxins that researcherswe have previously described that meets these requirements is aminochrome. Aminochrome is neurotoxic by being one-electron reduced to leukoaminochrome o-semiquinone radical by most flavoenzymes with the exception of DT-diaphorase (NADP(H):quinone oxidoreductase), which is the unique flavoenzyme that reduces quinones with two electrons [9][53]. In studies performed with electron spin resonance (ESR), NADPH cytochrome P450 reductase was found to reduce both dopamine ortho-quinone and aminochrome to the free radicals dopamine o-semiquinone and leukoaminochrome o-semiquinone, respectively. The ESR spectrum of dopamine o-semiquinone and of leukoaminochrome o-semiquinone radical was detected at 1 min. However, at 2 min only the dopamine o-semiquinone radical spectrum was detectable by ESR due to the high reactivity of leukoaminochrome o-semiquinone radical with oxygen [10]. Aminochrome one-electron reduction to the leukoaminochrome o-semiquinone radical induces oxidative stress due to the extremely rapid reaction with oxygen, which causes re-oxidation to aminochrome. This generates a redox cycling between aminochrome and leukoaminochrome o-semiquinone radical that functions until cellular dioxygen and/or NAD(P)H are depleted. This redox cycling generates oxidative stress and the depletion of cytosolic NADH for its use to generate ATP in the mitochondrial electron transport chain (Figure 1).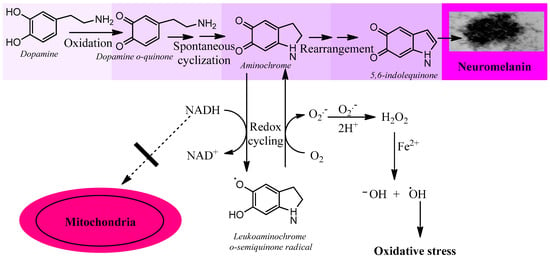
Figure 1. Aminochrome induces oxidative stress. Aminochrome can be reduced with one electron to produce the leukoaminochrome o-semiquinone radical by flavoenzymes using the cytosolic NADH generated to be used in the mitochondria to generate ATP through the transport chain coupled to oxidative phosphorylation from ADP to ATP. The leukoaminochrome o-semiquinone radical is extremely unstable in the presence of dioxygen and autoxidizes to regenerate aminochrome, which is reduced again, creating a redox cycle between aminochrome and leukoaminochrome o-semiquinone radical. This redox cycling is very fast and depletes as much oxygen as NADH with the concomitant production of oxidative stress.
3. Transcriptional Gene Activation via KEAP1/NRF2
Transcriptional activation of genes encoding a battery of cellular defense enzymes that provide protection against oxidative and electrophilic stress is affected by the transcription factor NRF2 (nuclear factor (erythroid-derived 2)-like 2) binding to an antioxidant responsive element or an electrophile responsive element (ARE/EpRE). Among the various cytoprotective proteins are γ-glutamylcysteine ligase, GSTs, and DT-diaphorase [29][30][18,64]. NFR2 is bound to KEAP1 (Kelch-like ECH-associated protein 1) in the cytosol, where it is directed to rapid proteasomal degradation. KEAP1 senses reactions of its sulfhydryl groups with electrophiles and oxidants and, as a consequence, NRF2 is released and targets ARE/ErRE elements of DNA in the nucleus. KEAP1/NRF2 is recognized as the most prominent protective system against electrophilic and oxidative insults; the upregulation of key enzymes in the defense against toxicants implicated in Parkinson’s disease and other neurodegenerative conditions is noteworthy. For obvious reasons, the KEAP1/NFR2 system has been considered for pharmacological interventions in Parkinson’s disease [31][32][19,65], Figure 2.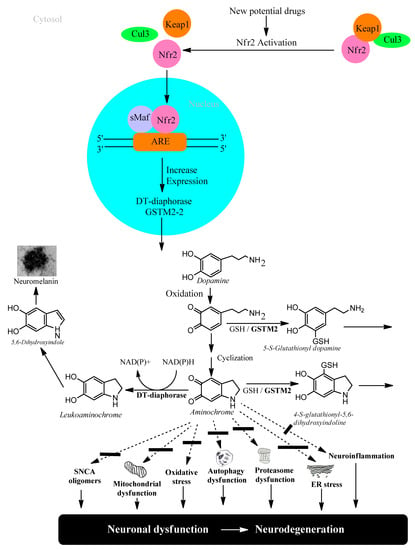
Figure 2. NFR2 activation to protect dopaminergic neurons from aminochrome neurotoxicity.
4. Neuroprotection against Neurodegeneration of Nigrostriatal System in Parkinson’s Disease
Studies carried out with postmortem material from healthy elderly people show intact neuromelanin-containing dopaminergic neurons, suggesting that neuromelanin synthesis is a natural and harmless process. Neuromelanin has been observed to increase over the years in the human brain [33][66]. However, there is an apparent contradiction, since the synthesis of neuromelanin requires the oxidation of dopamine to three ortho-quinones that can be neurotoxic. Among these aminochrome is the most toxic that induces oxidative stress, the formation of neurotoxic alpha-synuclein oligomers, dysfunction of both lysosomal and proteasomal protein breakdown systems, plasma reticulum stress, mitochondrial dysfunction, and neuroinflammation. The reason why the elderly have their neuromelanin-containing dopaminergic neurons intact at the time of death is because there are two enzymes that prevent the neurotoxic effects of aminochrome DT-diaphorase and glutathione transferase M2-2.4.1. DT-Diaphorase
DT-diaphorase (NAD(P)H: quinone oxidoreductase, NQO1) is a flavoenzyme with FAD as a prosthetic group that transfers two electrons from either NADH or NADPH to quinones, thereby reducing them to hydroquinones [9][26][34][53,61,67]. This enzyme is 90% located in the cytosol and 5% associated with the endoplasmic reticulum and mitochondria. It is widely expressed in different organs; in the brain it is expressed in the substantia nigra, striatum, cortex, hypothalamus, and hippocampus. In rat substantia nigra, DT-diaphorase is responsible for 97% of the total quinone reductase activity. DT-diaphorase is expressed in tyrosine hydroxylase positive neurons and in astrocytes. DT-diaphorase reduces aminochrome to leukoaminochrome, preventing its neurotoxic effects in a catecholaminergic cell line [35][68], but DT-diaphorase also prevents aminochrome induced-toxicity in a human astrocyte cell line [36][69]. DT-diaphorase prevents aminochrome-induced oxidative stress, mitochondrial dysfunction, formation of neurotoxic alpha-synuclein oligomers, proteasome dysfunction, autophagy dysfunction, and lysosome dysfunction [11][12][16][17][19][21][27][30][37][38][39][40][11,26,34,40,47,48,49,51,54,56,62,64].4.2. Glutathione Transferase M2-2
Electrophilic compounds such as epoxides, alkenals, and quinones can react with GSH and thereby become inactivated and form glutathione conjugates suitable for export from the cytosol [41][42][12,70]. The reactions are catalyzed by 20-odd enzymes, which are differentially distributed in cells and tissues. In humans and other mammals, the GSTs have been grouped into membrane-bound (microsomal) and soluble (cytosolic) proteins [43][44][13,71]. The former are members of the MAPEG (membrane-associated proteins in eicosanoid and glutathione metabolism) family [45][46][14,72]. The soluble GSTs are dimeric proteins occurring in eight classes of homologous sequences, which are catalytically active as homodimers as well as heterodimers [47][48][15,73]. The dimers are thus denoted according to their subunit composition, for example, as GST M1-1, GST M1-2, and GST M2-2 encoded by the GSTM1 and GSTM2 genes of the Mu class. With respect to the large variety of compounds identified as substrates for the various GSTs, it is noteworthy that only GST M2-2 has been found to catalyze with high efficiency the conjugation of ortho-quinones derived from dopamine [49][50][51][17,74,75]. The most active glutathione transferase catalyzing aminochrome conjugation is glutathione transferase M2-2, which gives rise to 4-S-glutathionyl-5,6-dihydroxyindoline. Interestingly, 4-S-glutathionyl-5,6-dihydroxyindoline is not oxidized by physiologically occurring oxidants such as dioxygen, hydrogen peroxide, and superoxide. Glutathione transferase M2-2 also catalyzes glutathione conjugation of the aminochrome precursor dopamine ortho-quinone to 5-glutathionyl dopamine that is degraded to 5-cysteinyl dopamine, which has been detected in human cerebrospinal fluid and neuromelanin [46][48][49][50][51][52][53][54][55][16,17,72,73,74,75,76,77,78] (Figure 3).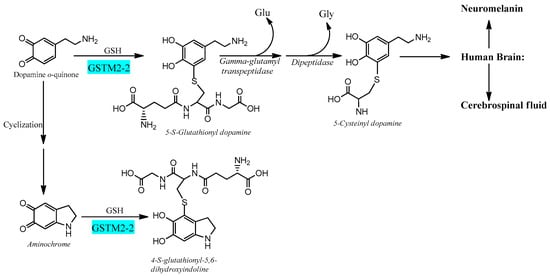
Figure 3. Glutathione transferase M2-2 catalyzes glutathione conjugation of ortho-quinones.
4.3. Astrocytes Protect Dopaminergic Neurons against Aminochrome Neurotoxicity
The brain is responsible for more than 20% of the energy consumption in the human body. The organism derives ATP via oxidation of carbohydrates, fats, and proteins and by coupling electron transport with phosphorylation of ADP to ATP. Neurons require ATP to transport proteins and neurotransmission vesicles to neuronal terminals, and neurotransmission itself is completely dependent on ATP. In other words, neuronal function is absolutely dependent on the presence of ATP, which, to a large extent, is generated in the mitochondria. Electrons in the transport chain from NADH flowing through complex I or complex II are further transmitted to ubiquinone Q10 (also known as coenzyme Q10), which in turn transfers the electrons to complex III. In this transfer of electrons, ubiquinone Q10 is reduced with one electron to the free radical ubiquinone Q10 semiquinone. The latter is subsequently reduced to ubiquinone Q10 hydroquinone, which transfers the electrons to complex III. However, the radical ubiquinone Q10 semiquinone is able to reduce dioxygen to superoxide, generating electron transport leakage and oxidative stress. Superoxide enzymatically or spontaneously can generate hydrogen peroxide, which, in the presence of reduced iron, forms hydroxyl radicals. This leakage of electrons from the mitochondrial electron transport chain permanently exposes neurons to conditions of oxidative stress. Astrocytes play an important role in protecting neurons against oxidative stress since astrocytes secrete precursors of glutathione, which is an important antioxidant [54][57][58][77,80,81]. Neuromelanin-containing dopaminergic neurons are also exposed to the neurotoxic effects of aminochrome, leading to oxidative stress, mitochondrial dysfunction, formation of neurotoxic alpha-synuclein oligomers, dysfunction of both lysosomal and proteasomal protein degradation systems, neuroinflammation, and endoplasmic reticulum stress [14][15][16][17][20][21][27][28][44,45,46,47,48,49,62,63]. However, astrocytes also play a neuroprotective role for neuromelanin-containing dopaminergic neurons by secreting exosomes loaded with the enzyme glutathione transferase M2-2 that penetrate dopaminergic neurons to, in concert with DT-diaphorase, prevent the neurotoxic effects of aminochrome [48][54][55][57][58][59][73,77,78,80,81,82] (Figure 4).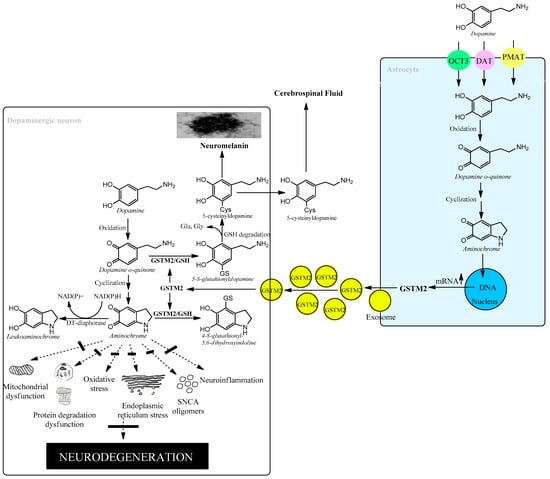
Figure 4. Astrocytes protect dopaminergic neurons against aminochrome neurotoxicity.