 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Francesco Giovinazzo | -- | 1934 | 2023-05-29 12:32:27 | | | |
| 2 | Rita Xu | Meta information modification | 1934 | 2023-05-30 03:35:51 | | |
Video Upload Options
Cholangiocarcinoma (CCA) encompasses all malignant neoplasms arising from the epithelial cells of the biliary tree. About 40% of CCAs are perihilar, involving the bile ducts distal to the second-order biliary branches and proximal to the cystic duct implant. About two-thirds of pCCAs are considered unresectable at the time of diagnosis or exploration. When resective surgery is deemed unfeasible, liver transplantation (LT) could be an effective alternative. The overall survival rates after LT at 1 and 3 years are 91% and 81%, respectively. The overall five-year survival rate after transplantation is 73% (79% for patients with underlying PSC and 63% for de novo pCCA). Multicenter case series reported a 5-year disease-free survival rate of ~65%.
1. Introduction
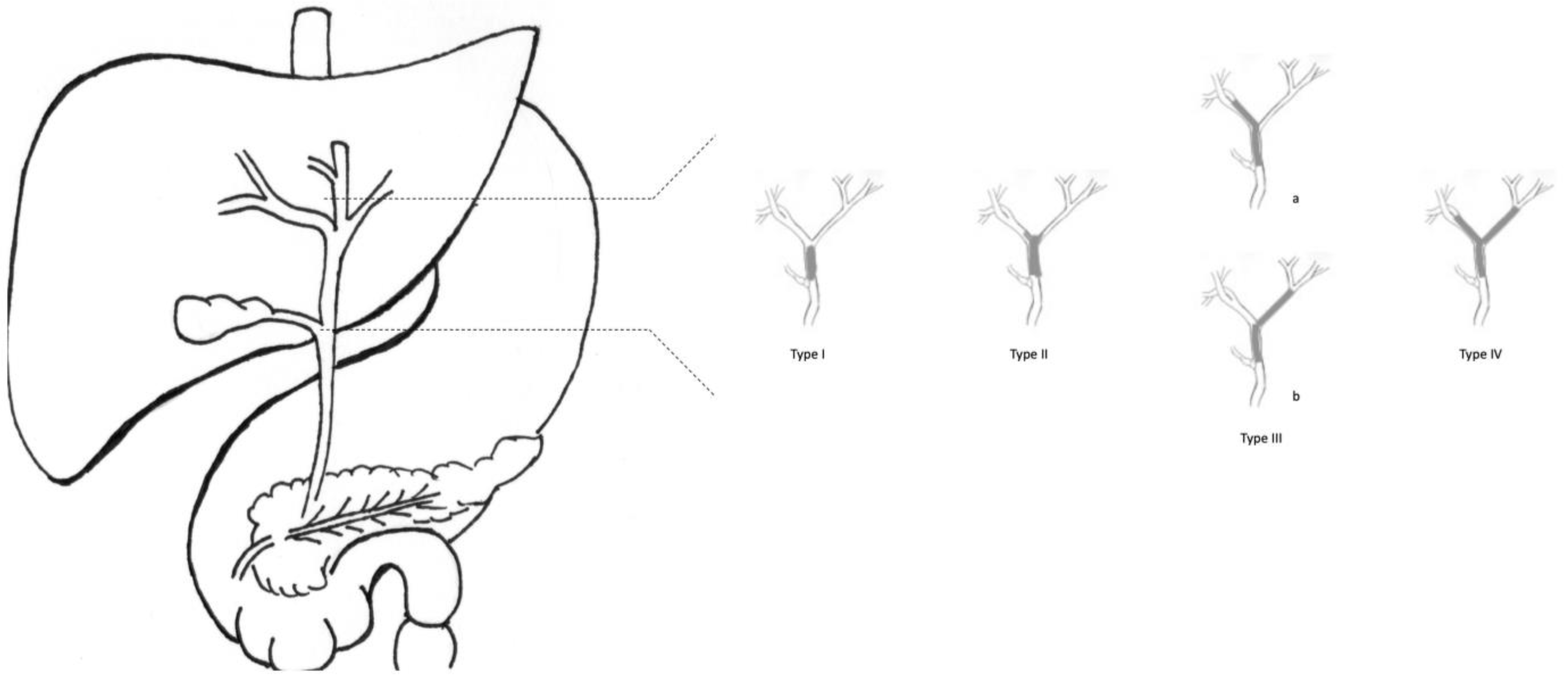
2. Results of Surgical Resection in pCCA
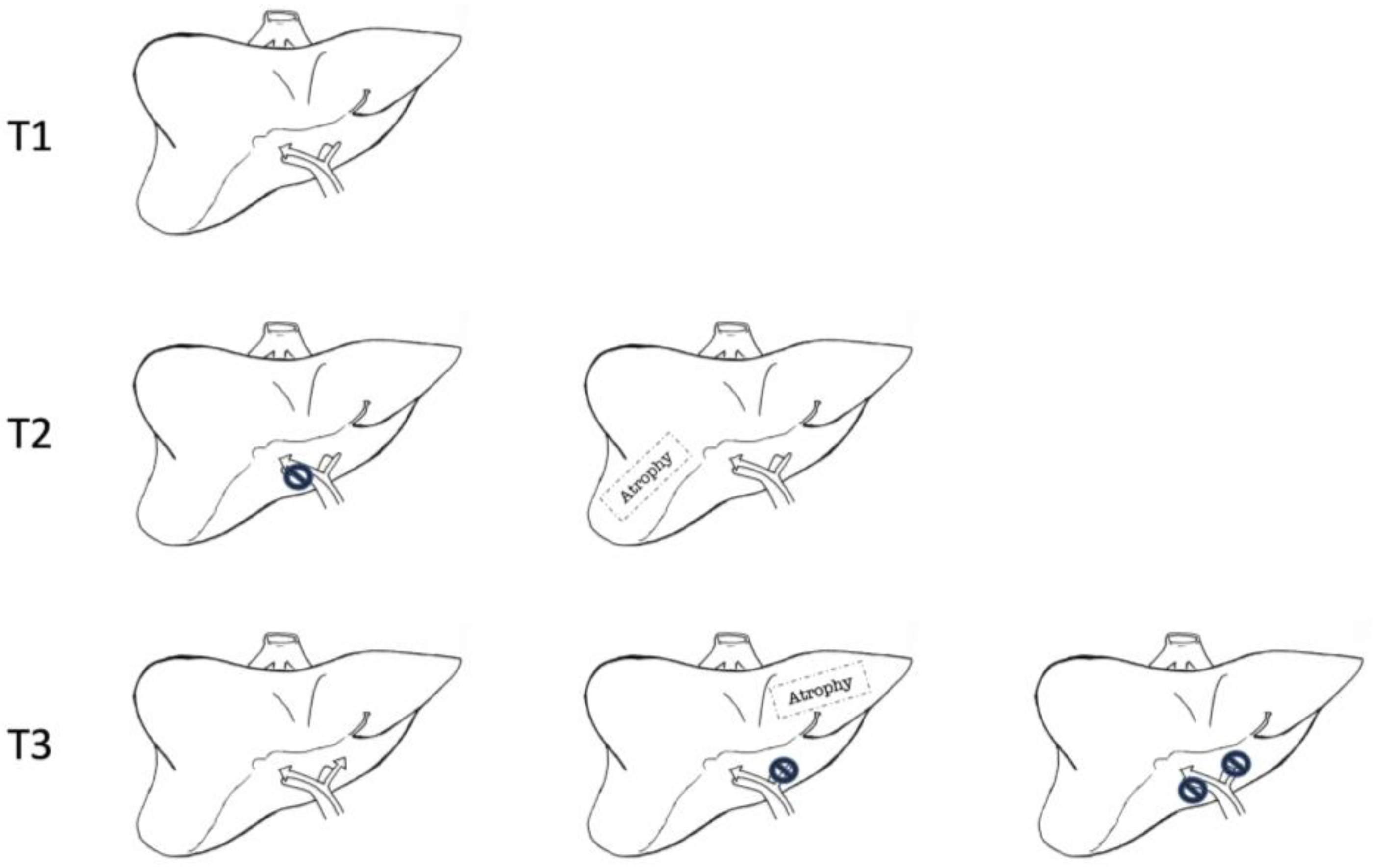
3. Liver Transplantation in pCCA
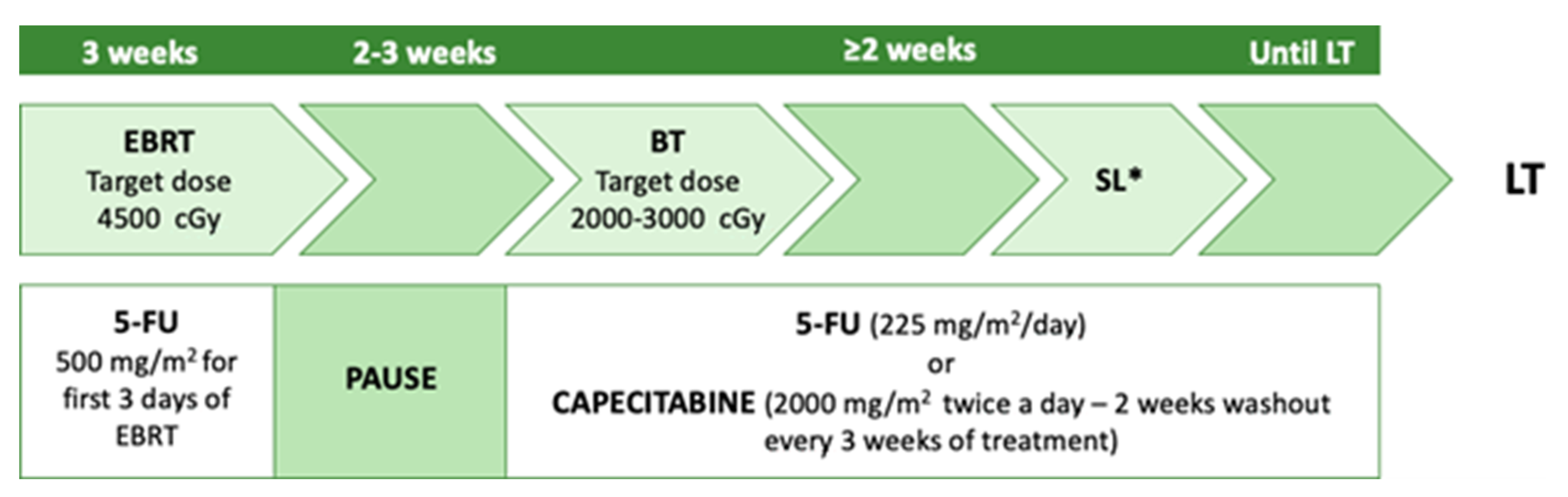
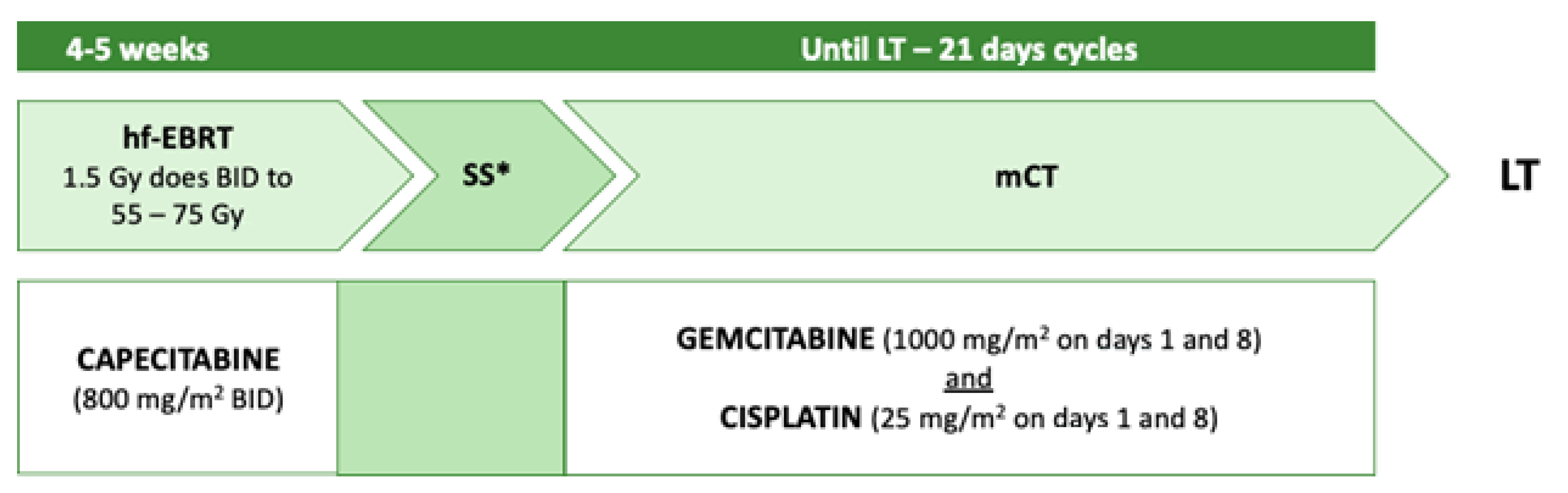
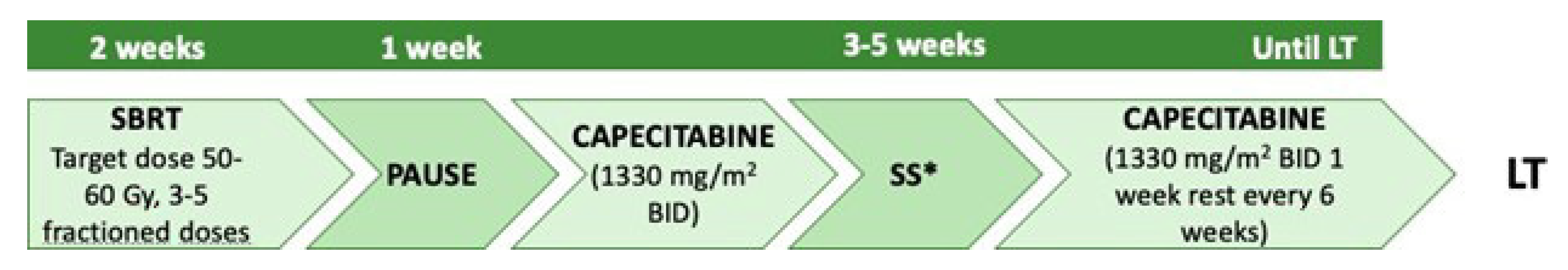
References
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179.
- Blechacz, B.; Komuta, M.; Roskams, T.; Gores, G.J. Clinical diagnosis and staging of cholangiocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 512–522.
- Gores, G.J. Cholangiocarcinoma: Current concepts and insights. Hepatology 2003, 37, 961–969.
- Khan, S.A.; Tavolari, S.; Brandi, G. Cholangiocarcinoma: Epidemiology and risk factors. Liver Int. 2019, 39, 19–31.
- DeOliveira, M.L.; Cunningham, S.C.; Cameron, J.L.; Kamangar, F.; Winter, J.M.; Lillemoe, K.D.; Choti, M.A.; Yeo, C.J.; Schulick, R.D. Cholangiocarcinoma: Thirty-one-year experience with 564 patients at a single institution. Ann. Surg. 2007, 245, 755–762.
- Launois, B.; Reding, R.; Lebeau, G.; Buard, J.L. Surgery for hilar cholangiocarcinoma: French experience in a collective survey of 552 extrahepatic bile duct cancers. J. Hepato-Biliary-Pancreat. Surg. 2000, 7, 128–134.
- Olthof, P.B.; Coelen, R.J.; Wiggers, J.K.; Koerkamp, B.G.; Malago, M.; Hernandez-Alejandro, R.; Topp, S.A.; Vivarelli, M.; Aldrighetti, L.A.; Campos, R.R. High mortality after ALPPS for perihilar cholangiocarcinoma: Case-control analysis including the first series from the international ALPPS registry. HPB 2017, 19, 381–387.
- Jarnagin, W.R.; Fong, Y.; DeMatteo, R.P.; Gonen, M.; Burke, E.C.; Bodniewicz, B.J.; Youssef, B.M.; Klimstra, D.; Blumgart, L.H. Staging, resectability, and outcome in 225 patients with hilar cholangiocarcinoma. Ann. Surg. 2001, 234, 507–519.
- Burke, E.C.; Jarnagin, W.R.; Hochwald, S.N.; Pisters, P.W.T.; Fong, Y.; Blumgart, L.H. Hilar cholangiocarcinoma: Patterns of spread, the importance of hepatic resection for curative operation, and a presurgical clinical staging system. Ann. Surg. 1998, 228, 385–394.
- Anderson, C.D.; Pinson, C.W.; Berlin, J.; Chari, R.S. Diagnosis and Treatment of Cholangiocarcinoma. Oncologist 2004, 9, 43–57.
- van Keulen, A.-M.; Olthof, P.B.; Cescon, M.; Guglielmi, A.; Jarnagin, W.R.; Nadalin, S.; Pratschke, J.; Ratti, F.; Troisi, R.I.; Koerkamp, B.G. Actual 10-Year Survival after Resection of Perihilar Cholangiocarcinoma: What Factors Preclude a Chance for Cure? Cancers 2021, 13, 6260.
- Cillo, U.; Fondevila, C.; Donadon, M.; Gringeri, E.; Mocchegiani, F.; Schlitt, H.J.; Ijzermans, J.N.M.; Vivarelli, M.; Zieniewicz, K.; Olde Damink, S.W.M. Surgery for cholangiocarcinoma. Liver Int. 2019, 39, 143–155.
- Vugts, J.J.A.; Gaspersz, M.P.; Roos, E.; Franken, L.C.; Olthof, P.B.; Coelen, R.J.S.; van Vugt, J.L.A.; Labeur, T.A.; Brouwer, L.; Besselink, M.G.H. Eligibility for Liver Transplantation in Patients with Perihilar Cholangiocarcinoma. Ann. Surg. Oncol. 2020, 28, 1483–1492.
- Meyer, C.G.; Penn, I.; James, L. Liver transplantation for cholangiocarcinoma: Results in 207 patients. Transplantation 2000, 69, 1633–1637.
- Kubal, C.; Mihaylov, P.; Holden, J. Oncologic indications of liver transplantation and deceased donor liver allocation in the United States. Curr. Opin. Organ Transplant. 2021, 26, 168–175.
- Heimbach, J.; Gores, G.; Haddock, M.; Alberts, S.; Nyberg, S.; Ishitani, M.; Rosen, C. Liver transplantation for unresectable perihilar cholangiocarcinoma. Semin. Liver Dis. 2004, 24, 201–207.
- Sudan, D.; DeRoover, A.; Chinnakotla, S.; Fox, I.; Shaw, B.J.r.; MCCAhland, T.; Sorrell, M.; Tempero, M.; Langnas, A. Radiochemotherapy and transplantation allow long-term survival for nonresectable hilar cholangiocarcinoma. Am. J. Transplant. 2002, 2, 774–779.
- Associazione Italiana di Oncologia Medica (AIOM). Linee Guida “Tumori delle Vie Biliari”. 2019. Available online: https://www.aiom.it/wp-content/uploads/2019/10/2019_LG_AIOM_Vie_biliari.pdf (accessed on 2 November 2022).
- Rosen, C.B.; Heimbach, J.K.; Gores, G.J. Liver transplantation for cholangiocarcinoma. Transpl. Int. 2010, 23, 692–697.
- Murad, S.D.; Kim, W.R.; Harnois, D.M.; Douglas, D.D.; Burton, J.; Kulik, L.M.; Botha, J.F.; Mezrich, J.D.; Chapman, W.C.; Schwartz, J.J.; et al. Efficacy of neoadjuvant chemoradiation, followed by liver transplantation, for perihilar cholangiocarcinoma at 12 US centers. Gastroenterology 2012, 143, 88–98.e3.
- Heimbach, J.K.; Haddock, M.G.; Alberts, S.R.; Nyberg, S.L.; Ishitani, M.B.; Rosen, C.B.; Gores, G.J. Transplantation for hilar cholangiocarcinoma. Liver Transplant. 2004, 10, S65–S68.
- Cambridge, W.A.; Fairfield, C.; Powell, J.J.; Harrison, E.M.; Søreide, K.; Wigmore, S.J.; Guest, R.V. Meta-analysis and Meta-regression of Survival After Liver Transplantation for Unresectable Perihilar Cholangiocarcinoma. Ann. Surg. 2021, 273, 240–250.
- Vogel, A.; Bridgewater, J.; Edeline, J.; Kelley, R.; Klümpen, H.; Malka, D.; Primrose, J.; Rimassa, L.; Stenzinger, A.; Valle, J.; et al. Biliary tract cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2023, 34, 127–140.
- Munir, M.M.; Ruff, S.M.; Endo, Y.; Lima, H.A.; Alaimo, L.; Moazzam, Z.; Shaikh, C.; Pawlik, T.M. Does Adjuvant Therapy Benefit Low-Risk Resectable Cholangiocarcinoma? An Evaluation of the NCCN Guidelines. J. Gastrointest. Surg. 2022.
- Loveday, B.P.T.; Knox, J.J.; Dawson, L.A.; Metser, U.; Brade, A.; Horgan, A.M.; Gallinger, S.; Greig, P.D.; Moulton, C.A. Neoadjuvant hyperfractionated chemoradiation and liver transplantation for unresectable perihilar cholangiocarcinoma in Canada. J. Surg. Oncol. 2018, 117, 213–219.
- Benson, A.B.; D’Angelica, M.I.; Abbott, D.E.; Anaya, D.A.; Anders, R.; Are, C.; Bachini, M.; Borad, M.; Brown, D.; Burgoyne, A.; et al. Hepatobiliary Cancers, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2021, 19, 541–565.
- Welling, T.H.; Feng, M.; Wan, S.; Hwang, S.Y.; Volk, M.L.; Lawrence, T.S.; Zalupski, M.M.; Sonnenday, C.J. Neoadjuvant stereotactic body radiation therapy, capecitabine, and liver transplantation for unresectable hilar cholangiocarcinoma. Liver Transplant. 2014, 20, 81–88.
- Duignan, S.; Maguire, D.; Ravichand, C.S.; Geoghegan, J.; Hoti, E.; Fennelly, D.; Armstrong, J.; Rock, K.; Mohan, H.; Traynor, O. Neoadjuvant chemoradiotherapy followed by liver transplantation for unresectable cholangiocarcinoma: A single-centre national experience. HPB 2014, 16, 91–98.
- Marchan, E.M.; Landry, J.C. Neoadjuvant chemoradiation followed by orthotopic liver transplantation in cholangiocarcinomas: The emory experience. J. Gastrointest. Oncol. 2016, 7, 248–254.


