 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Niloofar Piri | -- | 1998 | 2023-05-24 06:32:52 | | | |
| 2 | Rita Xu | Meta information modification | 1998 | 2023-05-24 07:29:24 | | |
Video Upload Options
Age-related macular degeneration (AMD) is a progressive degenerative disease of the central retina and the leading cause of severe loss of central vision in people over age 50. Patients gradually lose central visual acuity, compromising their ability to read, write, drive, and recognize faces, all of which greatly impact daily life activities. Quality of life is significantly affected in these patients, and there are worse levels of depression as a result. AMD is a complex, multifactorial disease in which age and genetics, as well as environmental factors, all play a role in its development and progression. The mechanism by which these risk factors interact and converge towards AMD are not fully understood, and therefore, drug discovery is challenging, with no successful therapeutic attempt to prevent the development of this disease.
1. Introduction
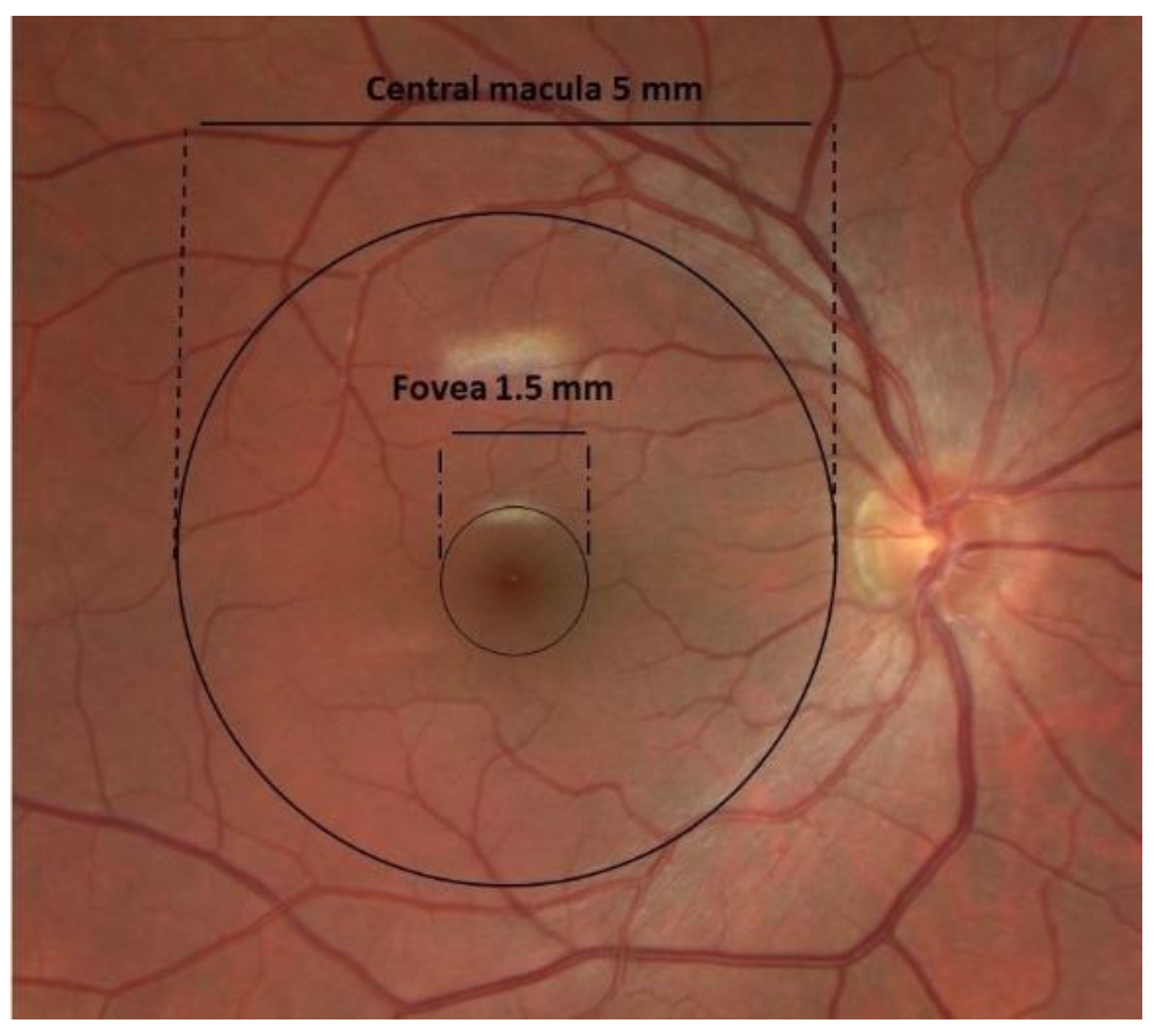
1.1. Anatomy of AMD
1.2. Pathophysiology of AMD
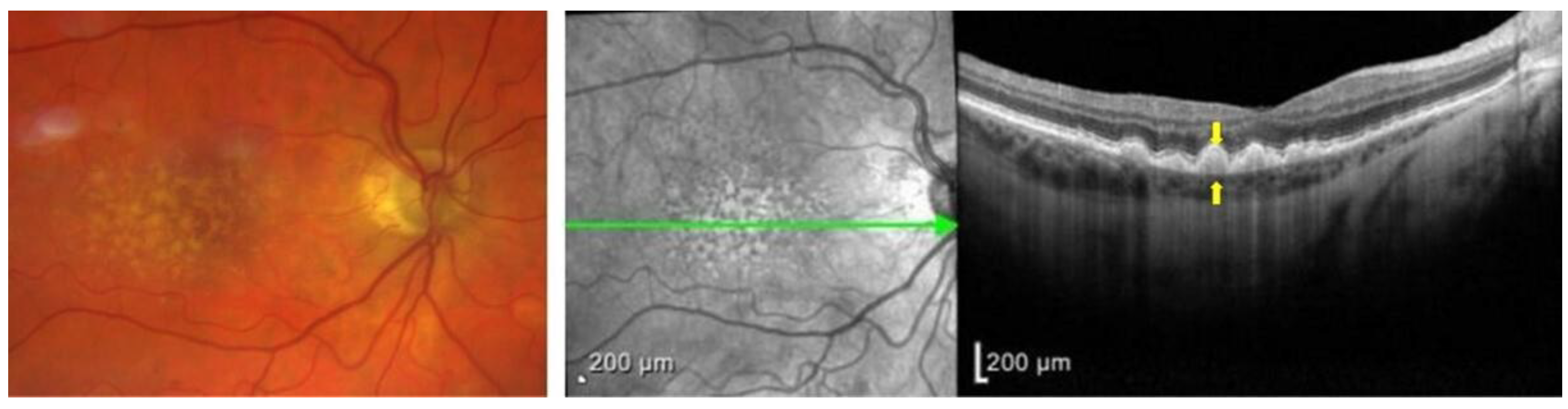
1.3. Clinical Presentation
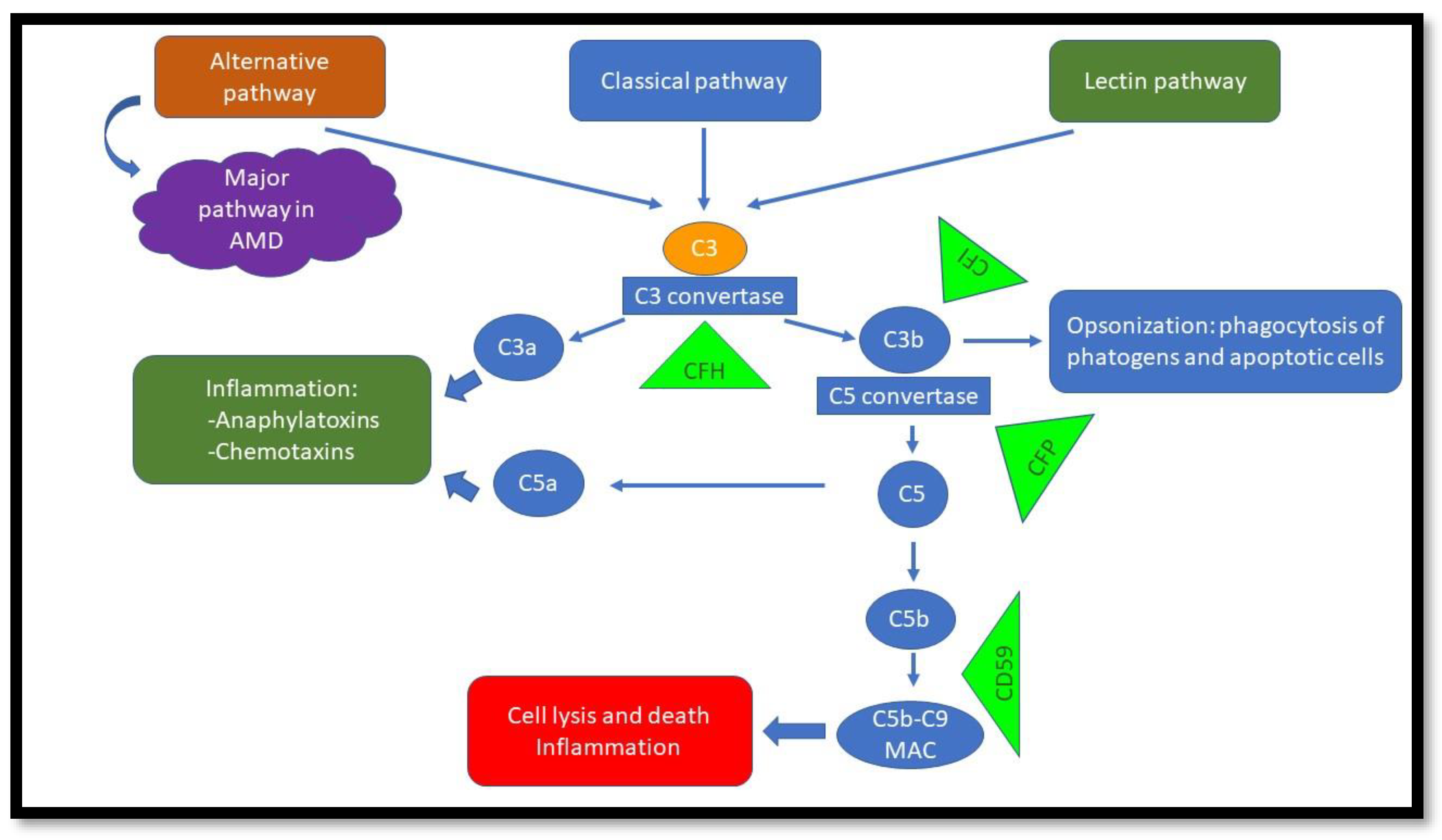
2. AMD and the Complement System
References
- Lim, L.S.; Mitchell, P.; Seddon, J.M.; Holz, F.G.; Wong, T.Y. Age-related macular degeneration. Lancet 2012, 379, 1728–1738.
- Rein, D.B.; Wittenborn, J.S.; Burke-Conte, Z.; Gulia, R.; Robalik, T.; Ehrlich, J.R.; Lundeen, E.A.; Flaxman, A.D. Prevalence of Age-Related Macular Degeneration in the US in 2019. JAMA Ophthalmol. 2022, 140, 1202–1208.
- Li, J.Q.; Welchowski, T.; Schmid, M.; Mauschitz, M.M.; Holz, F.G.; Finger, R.P. Prevalence and incidence of age-related macular degeneration in Europe: A systematic review and meta-analysis. Br J. Ophthalmol. 2020, 104, 1077–1084.
- Colijn, J.M.; Buitendijk, G.H.S.; Prokofyeva, E.; Alves, D.; Cachulo, M.L.; Khawaja, A.P.; Cougnard-Gregoire, A.; Merle, B.M.J.; Korb, C.; Erke, M.G.; et al. Prevalence of Age-Related Macular Degeneration in Europe: The Past and the Future. Ophthalmology 2017, 124, 1753–1763.
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106-116.
- Coleman, A.L.; Yu, F.; Ensrud, K.E.; Stone, K.L.; Cauley, J.A.; Pedula, K.L.; Hochberg, M.C.; Mangione, C.M. Impact of age-related macular degeneration on vision-specific quality of life: Follow-up from the 10-year and 15-year visits of the Study of Osteoporotic Fractures. Am. J. Ophthalmol. 2010, 150, 683–691.
- Coleman, H.R.; Chan, C.C.; Ferris, F.L., 3rd; Chew, E.Y. Age-related macular degeneration. Lancet 2008, 372, 1835–1845.
- Curcio, C.A.; Medeiros, N.E.; Millican, C.L. Photoreceptor loss in age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 1996, 37, 1236–1249.
- Chen, M.; Xu, H. Parainflammation, chronic inflammation, and age-related macular degeneration. J. Leukoc. Biol. 2015, 98, 713–725.
- Armento, A.; Ueffing, M.; Clark, S.J. The complement system in age-related macular degeneration. Cell. Mol. Life Sci. 2021, 78, 4487–4505.
- Ramrattan, R.S.; van der Schaft, T.L.; Mooy, C.M.; de Bruijn, W.C.; Mulder, P.G.; de Jong, P.T. Morphometric analysis of Bruch’s membrane, the choriocapillaris, and the choroid in aging. Invest. Ophthalmol. Vis. Sci. 1994, 35, 2857–2864.
- Hjelmeland, L.M.; Cristofolo, V.J.; Funk, W.; Rakoczy, E.; Katz, M.L. Senescence of the retinal pigment epithelium. Mol. Vis. 1999, 5, 33.
- Booij, J.C.; Baas, D.C.; Beisekeeva, J.; Gorgels, T.G.; Bergen, A.A. The dynamic nature of Bruch’s membrane. Prog. Retin. Eye Res. 2010, 29, 1–18.
- Feher, J.; Kovacs, I.; Artico, M.; Cavallotti, C.; Papale, A.; Balacco Gabrieli, C. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol. Aging 2006, 27, 983–993.
- Brown, E.E.; DeWeerd, A.J.; Ildefonso, C.J.; Lewin, A.S.; Ash, J.D. Mitochondrial oxidative stress in the retinal pigment epithelium (RPE) led to metabolic dysfunction in both the RPE and retinal photoreceptors. Redox Biol. 2019, 24, 101201.
- Wakatsuki, Y.; Shinojima, A.; Kawamura, A.; Yuzawa, M. Correlation of Aging and Segmental Choroidal Thickness Measurement using Swept Source Optical Coherence Tomography in Healthy Eyes. PLoS ONE 2015, 10, e0144156.
- Curcio, C.A.; Johnson, M.; Huang, J.D.; Rudolf, M. Aging, age-related macular degeneration, and the response-to-retention of apolipoprotein B-containing lipoproteins. Prog. Retin. Eye Res. 2009, 28, 393–422.
- Hageman, G.S.; Luthert, P.J.; Victor Chong, N.H.; Johnson, L.V.; Anderson, D.H.; Mullins, R.F. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog. Retin. Eye Res. 2001, 20, 705–732.
- Hageman, G.S.; Mullins, R.F. Molecular composition of drusen as related to substructural phenotype. Mol. Vis. 1999, 5, 28.
- Rudolf, M.; Clark, M.E.; Chimento, M.F.; Li, C.M.; Medeiros, N.E.; Curcio, C.A. Prevalence and morphology of druse types in the macula and periphery of eyes with age-related maculopathy. Invest. Ophthalmol. Vis. Sci. 2008, 49, 1200–1209.
- Mullins, R.F.; Russell, S.R.; Anderson, D.H.; Hageman, G.S. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. Faseb J. 2000, 14, 835–846.
- Klein, R.; Davis, M.D.; Magli, Y.L.; Segal, P.; Klein, B.E.; Hubbard, L. The Wisconsin age-related maculopathy grading system. Ophthalmology 1991, 98, 1128–1134.
- Miao, H.; Tao, Y.; Li, X.X. Inflammatory cytokines in aqueous humor of patients with choroidal neovascularization. Mol. Vis. 2012, 18, 574–580.
- Park, Y.G.; Park, Y.S.; Kim, I.B. Complement System and Potential Therapeutics in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2021, 22, 6851.
- Boyer, D.S.; Schmidt-Erfurth, U.; van Lookeren Campagne, M.; Henry, E.C.; Brittain, C. The Pathophysiology of Geographic Atrophy Secondary to Age-Related Macular Degeneration and the Complement Pathway as a Therapeutic Target. Retina 2017, 37, 819–835.
- Schramm, E.C.; Clark, S.J.; Triebwasser, M.P.; Raychaudhuri, S.; Seddon, J.; Atkinson, J.P. Genetic variants in the complement system predisposing to age-related macular degeneration: A review. Mol. Immunol. 2014, 61, 118–125.
- Morgan, B.P.; Walters, D.; Serna, M.; Bubeck, D. Terminal complexes of the complement system: New structural insights and their relevance to function. Immunol. Rev. 2016, 274, 141–151.
- Qin, S.; Dong, N.; Yang, M.; Wang, J.; Feng, X.; Wang, Y. Complement Inhibitors in Age-Related Macular Degeneration: A Potential Therapeutic Option. J. Immunol. Res. 2021, 2021, 9945725.
- Kim, B.J.; Mastellos, D.C.; Li, Y.; Dunaief, J.L.; Lambris, J.D. Targeting complement components C3 and C5 for the retina: Key concepts and lingering questions. Prog. Retin. Eye Res. 2021, 83, 100936.
- Harboe, M.; Ulvund, G.; Vien, L.; Fung, M.; Mollnes, T.E. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin. Exp. Immunol. 2004, 138, 439–446.
- Kim, S.J.; Kim, J.; Lee, J.; Cho, S.Y.; Kang, H.J.; Kim, K.Y.; Jin, D.K. Intravitreal human complement factor H in a rat model of laser-induced choroidal neovascularisation. Br J. Ophthalmol. 2013, 97, 367–370.
- Bora, N.S.; Jha, P.; Lyzogubov, V.V.; Kaliappan, S.; Liu, J.; Tytarenko, R.G.; Fraser, D.A.; Morgan, B.P.; Bora, P.S. Recombinant membrane-targeted form of CD59 inhibits the growth of choroidal neovascular complex in mice. J. Biol. Chem. 2010, 285, 33826–33833.
- Nozaki, M.; Raisler, B.J.; Sakurai, E.; Sarma, J.V.; Barnum, S.R.; Lambris, J.D.; Chen, Y.; Zhang, K.; Ambati, B.K.; Baffi, J.Z.; et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc. Natl. Acad. Sci. USA 2006, 103, 2328–2333.
- Lundh von Leithner, P.; Kam, J.H.; Bainbridge, J.; Catchpole, I.; Gough, G.; Coffey, P.; Jeffery, G. Complement factor h is critical in the maintenance of retinal perfusion. Am. J. Pathol. 2009, 175, 412–421.
- Rohrer, B.; Long, Q.; Coughlin, B.; Wilson, R.B.; Huang, Y.; Qiao, F.; Tang, P.H.; Kunchithapautham, K.; Gilkeson, G.S.; Tomlinson, S. A targeted inhibitor of the alternative complement pathway reduces angiogenesis in a mouse model of age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2009, 50, 3056–3064.
- Heesterbeek, T.J.; Lechanteur, Y.T.E.; Lorés-Motta, L.; Schick, T.; Daha, M.R.; Altay, L.; Liakopoulos, S.; Smailhodzic, D.; den Hollander, A.I.; Hoyng, C.B.; et al. Complement Activation Levels Are Related to Disease Stage in AMD. Invest. Ophthalmol. Vis. Sci. 2020, 61, 18.
- Merle, B.M.J.; Colijn, J.M.; Cougnard-Gregoire, A.; de Koning-Backus, A.P.M.; Delyfer, M.N.; Kiefte-de Jong, J.C.; Meester-Smoor, M.; Feart, C.; Verzijden, T.; Samieri, C.; et al. Mediterranean Diet and Incidence of Advanced Age-Related Macular Degeneration: The EYE-RISK Consortium. Ophthalmology 2019, 126, 381–390.
- Winkler, T.W.; Grassmann, F.; Brandl, C.; Kiel, C.; Günther, F.; Strunz, T.; Weidner, L.; Zimmermann, M.E.; Korb, C.A.; Poplawski, A.; et al. Genome-wide association meta-analysis for early age-related macular degeneration highlights novel loci and insights for advanced disease. BMC Med. Genom. 2020, 13, 120.
- Mullins, R.F.; Schoo, D.P.; Sohn, E.H.; Flamme-Wiese, M.J.; Workamelahu, G.; Johnston, R.M.; Wang, K.; Tucker, B.A.; Stone, E.M. The membrane attack complex in aging human choriocapillaris: Relationship to macular degeneration and choroidal thinning. Am. J. Pathol. 2014, 184, 3142–3153.
- Keenan, T.D.; Toso, M.; Pappas, C.; Nichols, L.; Bishop, P.N.; Hageman, G.S. Assessment of Proteins Associated With Complement Activation and Inflammation in Maculae of Human Donors Homozygous Risk at Chromosome 1 CFH-to-F13B. Invest. Ophthalmol. Vis. Sci. 2015, 56, 4870–4879.
- Whitmore, S.S.; Sohn, E.H.; Chirco, K.R.; Drack, A.V.; Stone, E.M.; Tucker, B.A.; Mullins, R.F. Complement activation and choriocapillaris loss in early AMD: Implications for pathophysiology and therapy. Prog. Retin. Eye Res. 2015, 45, 1–29.
- Sohn, J.H.; Kaplan, H.J.; Suk, H.J.; Bora, P.S.; Bora, N.S. Chronic low level complement activation within the eye is controlled by intraocular complement regulatory proteins. Invest. Ophthalmol. Vis. Sci. 2000, 41, 3492–3502.
- Lohman, R.-J.; Hamidon, J.K.; Reid, R.C.; Rowley, J.A.; Yau, M.-K.; Halili, M.A.; Nielsen, D.S.; Lim, J.; Wu, K.-C.; Loh, Z.; et al. Exploiting a novel conformational switch to control innate immunity mediated by complement protein C3a. Nat. Commun. 2017, 8, 351.
- Landowski, M.; Kelly, U.; Klingeborn, M.; Groelle, M.; Ding, J.D.; Grigsby, D.; Bowes Rickman, C. Human complement factor H Y402H polymorphism causes an age-related macular degeneration phenotype and lipoprotein dysregulation in mice. Proc. Natl. Acad. Sci. USA 2019, 116, 3703–3711.
- Acar İ, E.; Lores-Motta, L.; Colijn, J.M.; Meester-Smoor, M.A.; Verzijden, T.; Cougnard-Gregoire, A.; Ajana, S.; Merle, B.M.J.; de Breuk, A.; Heesterbeek, T.J.; et al. Integrating Metabolomics, Genomics, and Disease Pathways in Age-Related Macular Degeneration: The EYE-RISK Consortium. Ophthalmology 2020, 127, 1693–1709.
- Zhang, Y.; Gordon, S.M.; Xi, H.; Choi, S.; Paz, M.A.; Sun, R.; Yang, W.; Saredy, J.; Khan, M.; Remaley, A.T.; et al. HDL subclass proteomic analysis and functional implication of protein dynamic change during HDL maturation. Redox Biol. 2019, 24, 101222.
- Shaw, P.X.; Zhang, L.; Zhang, M.; Du, H.; Zhao, L.; Lee, C.; Grob, S.; Lim, S.L.; Hughes, G.; Lee, J.; et al. Complement factor H genotypes impact risk of age-related macular degeneration by interaction with oxidized phospholipids. Proc. Natl. Acad. Sci. USA 2012, 109, 13757–13762.
- Ferrington, D.A.; Ebeling, M.C.; Kapphahn, R.J.; Terluk, M.R.; Fisher, C.R.; Polanco, J.R.; Roehrich, H.; Leary, M.M.; Geng, Z.; Dutton, J.R.; et al. Altered bioenergetics and enhanced resistance to oxidative stress in human retinal pigment epithelial cells from donors with age-related macular degeneration. Redox Biol. 2017, 13, 255–265.
- Sivapathasuntharam, C.; Hayes, M.J.; Shinhmar, H.; Kam, J.H.; Sivaprasad, S.; Jeffery, G. Complement factor H regulates retinal development and its absence may establish a footprint for age related macular degeneration. Sci. Rep. 2019, 9, 1082.
- Ferrington, D.A.; Kapphahn, R.J.; Leary, M.M.; Atilano, S.R.; Terluk, M.R.; Karunadharma, P.; Chen, G.K.; Ratnapriya, R.; Swaroop, A.; Montezuma, S.R.; et al. Increased retinal mtDNA damage in the CFH variant associated with age-related macular degeneration. Exp. Eye Res. 2016, 145, 269–277.
- Nashine, S.; Chwa, M.; Kazemian, M.; Thaker, K.; Lu, S.; Nesburn, A.; Kuppermann, B.D.; Kenney, M.C. Differential Expression of Complement Markers in Normal and AMD Transmitochondrial Cybrids. PLoS One 2016, 11, e0159828.

