Age-related macular degeneration (AMD) is a progressive degenerative disease of the central retina and the leading cause of severe loss of central vision in people over age 50. Patients gradually lose central visual acuity, compromising their ability to read, write, drive, and recognize faces, all of which greatly impact daily life activities. Quality of life is significantly affected in these patients, and there are worse levels of depression as a result. AMD is a complex, multifactorial disease in which age and genetics, as well as environmental factors, all play a role in its development and progression. The mechanism by which these risk factors interact and converge towards AMD are not fully understood, and therefore, drug discovery is challenging, with no successful therapeutic attempt to prevent the development of this disease.
1. Introduction
Age-related macular degeneration (AMD) is the leading cause of severe loss of central vision in people over age 50
[1]. It is estimated that nearly 20 million Americans have some form of AMD
[2]. Currently, over 200 million people are affected worldwide, and it is estimated that this number will double by 2040
[3][4][5][3,4,5]. In Western countries, nearly 22% of people over 70 and 34% over 80 suffer from AMD in at least one eye
[5]. Patients gradually lose central visual acuity, compromising their ability to read, write, drive, and recognize faces, all of which greatly impair their daily life activities
[6].
1.1. Anatomy of AMD
The pathology of AMD is restricted to the outer retinal layers and the retinal pigment epithelium (RPE) of the macula
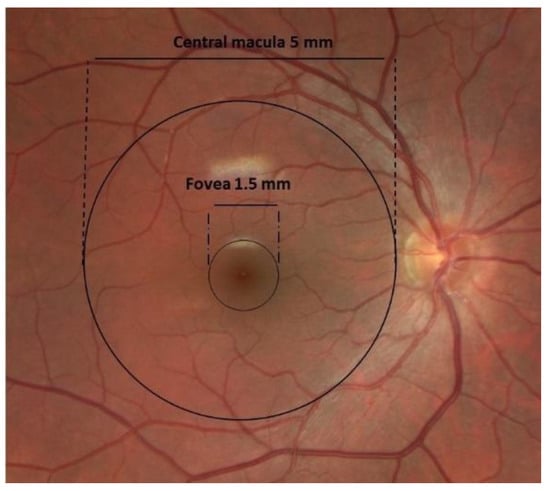
[7][9]. The central macula, or central posterior retina, measures 5 mm in diameter, with the highest density of cone photoreceptors located in the fovea, a 1500-micron area responsible for central visual acuity and color vision
[8][10] (
Figure 1). The fovea has a unique anatomical structure, both because of the high density of cone photoreceptors and the absence of retinal blood vessels
[9][11]. The cells in the fovea receive nutrition from the underlying choroidal circulation and the adjacent perifoveal capillary network. The absence of an inner retinal vasculature allows improved light absorption by the cone photoreceptor cells, providing high-resolution vision
[10][12].
Figure 1. Clinical fundus photograph of the macula and fovea and their dimensions.
1.2. Pathophysiology of AMD
Major changes in BM contribute to macular damage, including increased thickness, which is a result of increased deposition and the cross-linking of collagen fibers, and decreased permeability, resulting in decreased RPE metabolic function
[10][11][12][12,16,17]. It has also been observed that with aging, there is a deposition of advanced glycated end products (AGEs), i.e., glycated or oxidized lipoproteins
[13][18], which can trigger inflammation, as well as the presence of leukocytes in the choroid underneath the macula in AMD.
RPE cells lose melanosomes, and mitochondria become more prone to oxidative stress with aging
[14][15][19,20]. This is associated with major changes in the choroid. The normal choroid has high blood flow and can change rapidly, depending on the physiologic demand of the RPE and neurosensory retina. Aging leads to decreased blood flow, as well as the thinning of the choroid
[16][21]. Therefore, flexibility decreases, and the choroid cannot rapidly respond to changes in blood flow required for the physiologic demand. Decreased blood flow results in a decreased support of oxygen and nutrients to the retina. The resultant hypoxia and lack of sufficient nutrient support drives RPE cells to extreme metabolic stress. Additionally, reduced blood flow impairs the removal of photoreceptor and RPE waste products, which are now deposited in the BM, leading to drusen formation
[17][22].
“Drusen” are extracellular deposits between the basal lamina of the RPE and the inner collagenous layer of BM
[18][19][23,24]. They consist of many different components, and the composition varies with the disease stage. More than 40% of its contents include neutral lipids and esterified and unesterified cholesterol
[17][22]. In addition, more than 129 proteins have been identified in drusen
[20][25], including inflammatory and complement factors, such as vitronectin, serum beta amyloid protein, tissue inhibitor of metalloproteinase 3 (TIMP3), apolipoproteins (E, B, A-I, C-I, and C-II), immunoglobulin light chains, complement proteins (C5 and the C5b-9 complex), proteins involved in complement regulation, and zinc and iron ions
[17][20][21][22,25,26].
1.3. Clinical Presentation
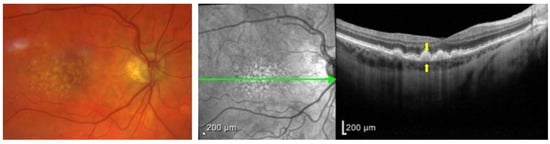
Upon clinical examination of the fundus, drusen present as small, yellowish-white spots underneath the retina in the central macula. They are classified clinically in the Wisconsin grading system as hard or soft drusen. Hard drusen are defined as well-defined yellow spots that are 1–63 microns in diameter. Soft drusen are larger than 125 microns, or if they are 63–125 microns, they have visible thickness. Soft drusen can have either distinct or indistinct borders (
Figure 2)
[22][27]. They are linked with a higher risk of choroidal neovascularization (CNV) because they include C3a and C5a and can trigger the overexpression of the vascular endothelial growth factor (VEGF) in RPE
[23][28].
Figure 2. (Left) Color fundus photograph of a patient with intermediate AMD demonstrating soft drusen, with indistinct borders. (Right) Optical coherence tomography (OCT) imaging of the central macula in the same patient demonstrating an optical section of all retinal layers. Notice the hyperreflective deposits between the RPE (top arrow)and the BM (bottom arrow), which represent drusen from the left panel.
2. AMD and the Complement System
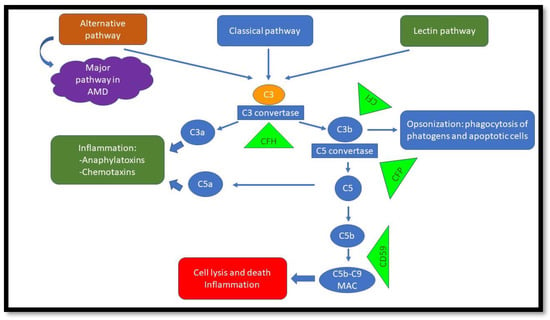
The complement system is a crucial component of the innate immune response that protects the host against invading pathogens, such as bacteria. It has three pathways: classic (antigen–antibody complex), lectin-dependent (mannose polysaccharides on microorganisms), and alternative (pathogen cell surfaces)
[24][58]. Each pathway has its own distinctive factors, but they all converge and ultimately lead to the cleavage of complement factor 3 (C3) to C3a and C3b. C3a is anaphylatoxin and induces inflammation, and C3b opsonizes cells and labels them for phagocytosis. The complement cascade continues and ultimately forms the membrane attack complex (MAC), which includes (C5b-C9). The MAC penetrates the cell membrane and results in cell death
[24][25][26][58,59,60]. The formation of the MAC can be inhibited by complement inhibitors, such as CD59
[27][61]. Under normal conditions, host cell regulatory proteins deactivate active complement components and prevent damage to host cells
[25][59](
Figure 3).
Figure 3. Complement pathways. The alternative pathway is important in AMD. Green triangles demonstrate regulators/inhibitors of the specific pathways. Malfunction will lead to overactivation of complement and, ultimately, cell damage.
The complement system can be considered a double-edged sword. On one side, under normal conditions, it protects the body against foreign pathogens and modifies the surveillance of immune responses; on the other side, under abnormal conditions, its dysregulation or dysfunction can lead to uncontrolled inflammatory responses and can damage host tissue, resulting in disease
[28][62].
The alternative pathway is activated in two different ways. The first one is called “tickover”, which is the spontaneous hydrolysis of C3 into C3 (H2O) in the fluid phase. The second one is by C4b2a (C3 convertase), which results in the cleavage of C3 to C3a and C3b in the solid phase
[29][63]. The alternative pathway is responsible for the majority (80–90%) of the terminal pathway activation
[30][64]; therefore, overactivation of this pathway may have a major role in disease pathogenesis. Early investigations initially debated whether systemic or local complement activation caused disease in the eye. However, it may well be that complement-mediated molecular processes causing AMD are a combination of systemic complement proteins, as well as locally synthesized complement proteins
[10][12]. Experimental studies in animal models suggested the idea above.
The most commonly used animal model mimics wet AMD and is induced by retinal laser photocoagulation in rodents. Laser-induced CNV can be suppressed by blocking complement activation via local or systemic mechanisms. Inhibiting C3a, C5a, CFB, and MAC or administering the complement regulating molecules CD59 and CFH can prevent the formation of CNV in animal models
[31][32][33][65,66,67]. In CFH-deficient mice, excessive alternative pathway activation resulted in the enlargement of the induced CNV
[34][68]. It was also shown that mice lacking the alternative complement pathway (CFB-deficient) had decreased rates of CNV, compared to animals lacking other complement pathway constituents
[33][35][67,69].
In individuals with AMD, several complement system activation products have been identified to be increased systemically
[36][70]. Patients with intermediate or late dry AMD (i.e., central GA or inactive CNV) have higher levels of systemic complement activation, compared to both controls and early AMD
[10][12]. In contrast, the level of systemic complement activation in wet AMD is relatively low
[10][36][12,70]. Study of donor eyes from patients with the CFH gene showed increased deposits of local C3b before clinical manifestations of the disease. Thus, poorly controlled complement turnover may start earlier than clinical disease manifestation within the retina
[36][70].
Five AMD risk alleles have been recognized involving the alternative complement pathway, implying a role for innate immunity in disease development. Additionally, the pathogenesis of early AMD may be different than that of late-stage AMD
[37][35]. A recent GWAS identified a gene variant near CD46 almost exclusively associated with early AMD
[38][71].
Interestingly, complement products and the membrane attack complex (MAC) have been found in the choroids of eyes from young donors, as well as those without genetic risks for AMD. This suggests that low-grade complement activation is likely required for the normal homeostasis of the retina/RPE complex
[10][39][40][41][12,72,73,74], similar to observations made in the anterior chamber of the eye
[42][75]. The complement system may assist in the removal of waste products, such as the shed photoreceptor outer segments. MAC levels increase with aging, possibly leading to bystander complement-mediated injury of the tissue
[10][39][40][41][12,72,73,74].
A frequent consequence of complement activation is the recruitment and activation of immune cells and the release of the anaphylatoxins C3a and C5a
[10][12]. The immune cells involved in AMD comprise resident microglial cells, as well as circulating lymphocytes, monocytes/macrophages, and mast cells. Stimulation of monocytes by C3a leads to the secretion of multiple cytokines, possibly leading to tissue damage. Mast cell degranulation by C3a and C5a can lead to the release of proteases, tryptase, and chymase, with resultant damage to the extracellular matrix (ECM)
[43][76].
Lipid deposition in the retina has also been linked to both systemic and local complement systems dysregulation
[44][77]. Studies on the serum of AMD patients demonstrated a substantial correlation between elevated large and extra-large high-density lipoprotein (HDL) levels and decreased, very low-density lipoprotein (VLDL) and amino acid levels. These changes were associated with elevated serum complement activation
[45][78].
CFH has been shown to be present in large HDL particles but not in small or medium HDL molecules
[46][79]. Whether this is pro-inflammatory or suppressive for inflammatory responses is not yet clear. Entrapment by HDL might decrease the availability of circulating CFH to deactivate the complement cascade
[46][79]. On the other hand, the presence of CFH in drusen might suppress inflammation locally in the retina. The latter has supportive evidence, since it has been shown that factor H binds the native low-density lipoprotein (LDL) and the oxidized LDL; this binding affinity is reduced in patients with the CFH high-risk 402H variant. As a result, increased levels of oxidized LDL may lead to the upregulation of inflammatory cytokines and the start of an inflammatory cycle that results in tissue damage
[47][80].
It was also observed that disturbed RPE metabolism is important in the pathogenesis of AMD. Compared to RPE cells from healthy controls, primary RPE cells isolated from AMD patients were shown to have severely impaired energy metabolism
[48][81].
Complement dysregulation associated with the CFH variant is associated with reduced energy metabolism in RPE cells
[49][82]. Complement dysregulation was associated with mitochondrial damage in RPE cells
[50][83]. Compared to RPE cells from donors with the low-risk 402Y genetic variation, RPE cells from AMD donors harboring the high-risk 402H variant exhibited greater mtDNA damage
[50][83]. In the CFH (cfh-/-) knock-out mouse model, abnormally large mitochondria were shown in the RPE and photoreceptors. At the same time, the amount of mitochondrial DNA (mt DNA) decreased, which is a sign of energy dysregulation confirmed by lower adenosine tri-phosphate (ATP) production
[49][82]. Regarding an oxidative stress response, there seems to be a reciprocal interaction between the complement system control and energy metabolism
[10][12]. In a cytoplasmic hybrid (cybrid) model, with mitochondria from AMD patients and ARPE19 devoid of mitochondria, changes in complement components were demonstrated. Complement activators, including CFB, were found to be increased, and complement inhibitors, including CFH, were found to be decreased
[51][84].