Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sang Ryong Kim | -- | 2335 | 2023-05-17 10:40:07 | | | |
| 2 | Rita Xu | Meta information modification | 2335 | 2023-05-17 11:06:07 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kim, S.; Sharma, C.; Jung, U.J.; Kim, S.R. Blood-Borne Proteins in Alzheimer’s Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/44420 (accessed on 26 June 2026).
Kim S, Sharma C, Jung UJ, Kim SR. Blood-Borne Proteins in Alzheimer’s Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/44420. Accessed June 26, 2026.
Kim, Sehwan, Chanchal Sharma, Un Ju Jung, Sang Ryong Kim. "Blood-Borne Proteins in Alzheimer’s Disease" Encyclopedia, https://encyclopedia.pub/entry/44420 (accessed June 26, 2026).
Kim, S., Sharma, C., Jung, U.J., & Kim, S.R. (2023, May 17). Blood-Borne Proteins in Alzheimer’s Disease. In Encyclopedia. https://encyclopedia.pub/entry/44420
Kim, Sehwan, et al. "Blood-Borne Proteins in Alzheimer’s Disease." Encyclopedia. Web. 17 May, 2023.
Copy Citation
The blood–brain barrier (BBB) restricts entry of neurotoxic plasma components, blood cells, and pathogens into the brain, leading to proper neuronal functioning. BBB impairment leads to blood-borne protein infiltration such as prothrombin, thrombin, prothrombin kringle-2, fibrinogen, fibrin, and other harmful substances. Thus, microglial activation and release of pro-inflammatory mediators commence, resulting in neuronal damage and leading to impaired cognition via neuroinflammatory responses, which are important features observed in the brain of Alzheimer’s disease (AD) patients.
Alzheimer’s disease
microglia
neuroinflammation
neurodegeneration
blood–brain barrier
1. Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder often characterized by memory loss and impaired cognition [1][2][3][4]. The global AD prevalence is increasing concurrently with the aging population [4][5][6][7]. However, the pathogenic mechanisms of AD remain elusive, despite extensive research on the various causative and contributory factors, including genetics [1][2][3][8][9][10][11][12]. An accumulating body of evidence suggests that neuroinflammation mediated by glial activation is an important neurotoxic mechanism associated with the initiation and progression of AD, including the subsequent neuronal cell death and cognitive impairment [13][14][15][16][17][18][19][20][21][22][23][24].
Microglia promote brain homeostasis through synaptic remodeling maintenance and dead cell removal under non-pathological conditions. However, during the early stages of AD, an increase in inflammatory responses via microglial activation in the brain has been reported [23][25][26]. These responses are also involved in the early formation of amyloid beta (Aβ) plaques and persistent microglial activation, which can result in the generation of pro-inflammatory cytokines and reactive oxygen species (ROS) [23][25][26]. These pathways interact and reinforce one another to induce pathological alterations in the AD brain, which primarily involve microglial activation, neuroinflammation, tau hyperphosphorylation, compromised mitochondrial activity, and oxidative stress [23][25][26]. This, in turn, triggers neurodegeneration due to the subsequent loss of synapses and neurons, resulting in neuronal death [14][15][17][18][20]. In particular, the initial microglial activation and the resulting neuroinflammation may act as a starting point for the observed neurodegeneration in the AD brain [23][25][26]. These findings suggest that initial microglial activation can exacerbate AD progression, despite potential variations in the roles of microglia activation and inflammatory responses within the brain. [13][17][18][27][28][29][30]. Therefore, there is a pressing need to investigate such endogenous substances that may induce microglial activation to build an effective AD treatment strategy.
The blood–brain barrier (BBB) is essential for the maintenance of the brain tissue microenvironment. It improves connectivity while also providing insulation of the brain parenchyma from the peripheral circulation system [31]. It is composed of specialized endothelial cells and various tight junction (TJ) proteins to ensure optimal functioning of the BBB [31]. These TJs, together with specialized transporters, allow the passage of nutrients into the central nervous system (CNS) while restricting blood-borne molecule influx [32]. Meanwhile, in pathological conditions, BBB disruption can result in the entry of blood-borne proteins such as prothrombin, thrombin, prothrombin kringle-2 (pKr-2), fibrinogen, and fibrin, among others, into the brain parenchyma [33][34]. This causes gradual neurodegeneration and neuronal death through direct and/or indirect neurotoxicity, oxidative stress, mitochondrial function loss, and pro-inflammatory mediator release, leading to memory loss and a decline in cognitive function. Several reports suggest that the early stages of AD are critical, as BBB breakdown occurs during this period [35][36][37][38][39], which allows the entry of blood-borne proteins in the brain parenchyma, thus contributing to neurotoxicity by several mechanisms [40][41]. For instance, blood-borne proteins such as prothrombin, thrombin, pKr-2, fibrinogen, and fibrin can activate microglia and induce neuroinflammation, resulting in neurodegeneration and cognitive decline in various animal models of AD [34][42][43][44][45][46][47][48][49][50][51]. Moreover, some of these blood proteins (such as thrombin, fibrinogen, and fibrin) can cluster with Aβ, the main causal agent of AD, and can further exacerbate microglial activation [52][53][54][55][56]. Furthermore, fibrinogen deposition can lead to impaired memory and result in exacerbated levels of interleukin-6, ROS, mitochondrial superoxide, and nitrite in mouse brain neurons [57]. This can result in abnormal tau phosphorylation, which is another major hallmark of AD [58], and further amplify neurotoxicity through the interplay with Aβ plaques, thus affecting neurotransmitter balance [59]. As fibrinogen is not expressed in the brain, BBB disruption is required for plasma fibrinogen to cross the BBB and reach the brain parenchyma [34]. Therefore, investigating the mechanisms of blood-borne protein entry into the brain and microglial activation promotion leading to the release of pro-inflammatory cytokines resulting in neuronal loss may help develop novel therapeutic strategies for AD.
2. Blood-Borne Protein by BBB Leakage Induces Microglial Activation in AD
The BBB prevents the entry of harmful substances into the brain from the blood [60][61][62][63]. Age-related degeneration of the BBB increases its permeability along with a decrease in cerebral blood flow [63][64][65][66][67]. A decrease in the interaction between neurons and endothelial cells and astrocytes is another factor that contributes to the BBB breakdown and impairment. This results in the blood-borne substance leakage into the brain, including prothrombin, thrombin, pKr-2, fibrinogen, fibrin, and albumin (Figure 1) [34][46][51][68].
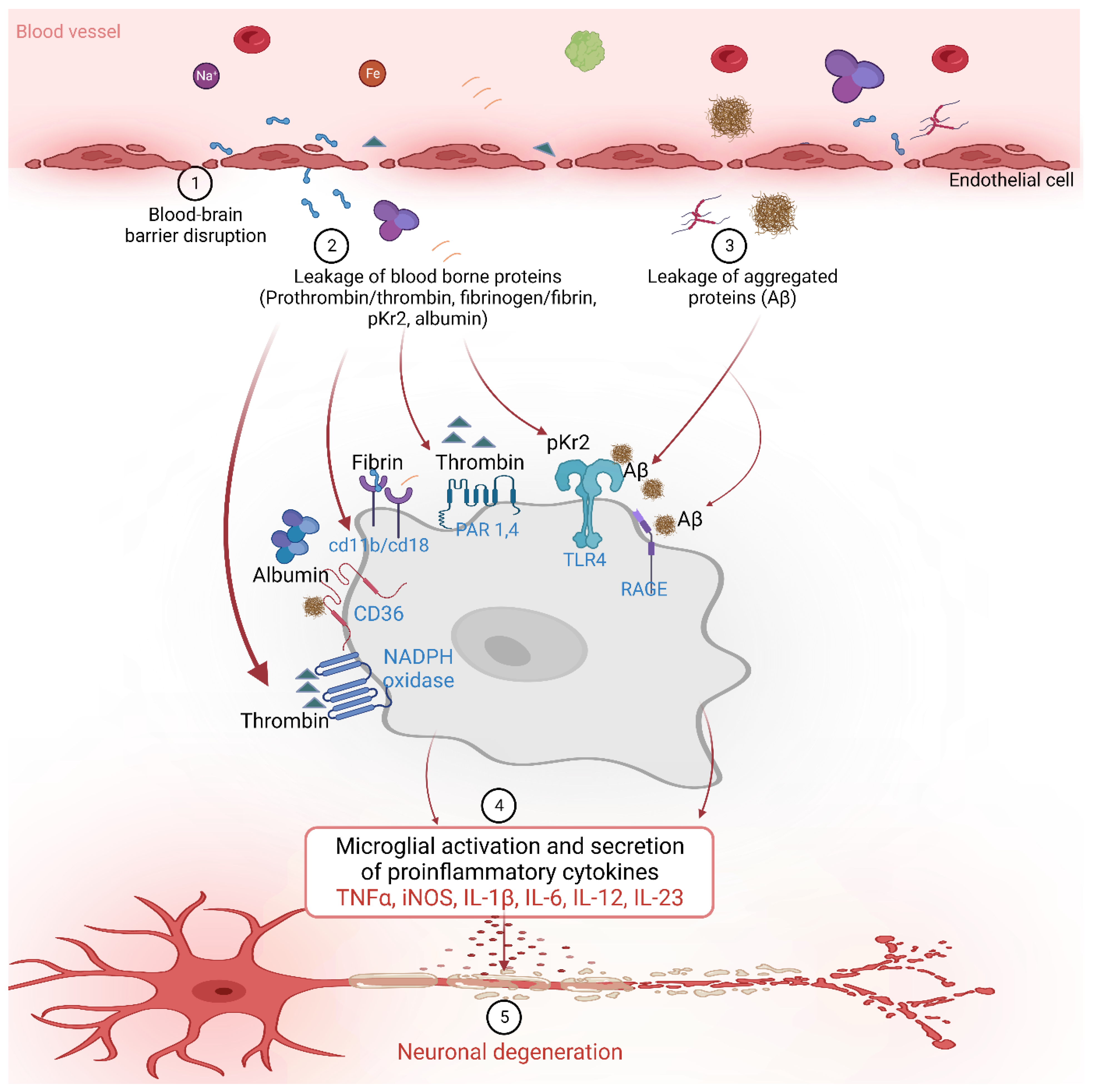
Figure 1. The blood-borne proteins interact with various receptors on the microglial cells to activate downstream signaling and influence various inflammatory and neurodegenerative processes in Alzheimer’s disease.
2.1. Prothrombin and Thrombin
Prothrombin is a protein involved in the blood coagulation pathway, which helps to form blood clots in response to injury [69][70][71][72][73]. Prothrombin in blood plasma is converted into thrombin by factor X or prothrombinase, which then transforms fibrinogen into fibrin to form a clot with platelets, preventing blood loss [69][70][71][72][73]. Increased expression of prothrombin has been found in both neurons and glial cells in the brain tissue of patients with AD patients compared with healthy controls [48][50][74][75][76]. Furthermore, prothrombin has been shown to accumulate in neurofibrillary tangles, a pathological hallmark of AD [48]. Although the precise mechanisms underlying the upregulation of prothrombin and thrombin in AD are not completely understood, a growing body of evidence suggests their potential role in the pathogenesis of this neurodegenerative disease [48][49][50][51][74][75]. These findings suggest that prothrombin may contribute to the neurodegenerative process in AD, particularly concerning neuronal damage [48][74][76]. Thrombin is a protease that is also involved in the blood coagulation pathway, but it can also be generated in the brain in response to injury and inflammation [77][78][79]. In AD, thrombin is upregulated in the brain and may contribute to the pathogenesis of the disease [48][50][74]. In one study, the expression of prothrombin mRNA in brain tissues, neuroblastoma cells, cultured human astrocytes, oligodendrocytes, and microglial cells were detected using the reverse transcriptase–polymerase chain reaction, and the presence of prothrombin and thrombin proteins in the brain was confirmed using specific monoclonal and polyclonal antibodies for both proteins through immunohistochemistry [48]. Furthermore, studies have demonstrated increased expression of thrombin and its receptors such as protease-activated receptor 1 (PAR-1) and protease-activated receptor 4 (PAR-4) in the brain in neurodegenerative diseases [74][80]. Thrombin has also been reported to induce the NADPH oxidase in microglia in the hippocampus, leading to the death of neuronal cells, suggesting a potential mechanism by which thrombin contributes to neurodegenerative processes in the brain [49]. Other reports also show that thrombin can activate microglia, leading to pro-inflammatory cytokine releases such as TNF-α and IL-1β in BV-2 and primary human microglia in vitro [81][82].
Thrombin-induced neuroinflammation via microglial activation was found to be mediated through the PAR-1 receptor signaling pathway [80]. In the AD brain, thrombin can also induce vascular inflammation, exacerbating BBB damage [50][51]. Thrombin may also contribute to Aβ accumulation in the brain, as it can cleave the Aβ precursor protein and generate Aβ peptides that aggregate and form amyloid plaques in the cultured hippocampal neurons and human endothelial cells in vitro [52][53]. Additionally, reports suggest that thrombin can induce tau hyperphosphorylation and aggregation in murine hippocampal neurons, resulting in microglial activation, reduction in delayed synaptophysin, and apoptotic neuronal death [83]. Moreover, along with calpain-1, it can cleave tau, resulting in a ~17 kDA neurotoxic fragment that can cause impaired synaptic function and axonal transport, mitochondrial membrane depolarization, and behavioral deficits in AD brains [84][85].
2.2. pKr-2
pKr-2, also known as fragment 2, is a small protein fragment that is released during the activation of prothrombin [44][45][46][47][86][87][88][89][90][91][92][93][94][95][96][97][98], which is a precursor protein that is converted into the active blood-clotting enzyme thrombin [44][45][46][47][86][87][88][89][90][91][92][93][94][95][96][97][98]. The role of pKr-2 in the blood is to inhibit the prothrombinase complex activity in conjunction with Factor Va, thereby preventing further thrombin production [45][95][99]. In addition, pKr-2 inhibits endothelial cell proliferation and angiogenesis, suggesting its potential role in BBB maintenance [91][92].
Researchers recently reported the increased pKr-2 protein expression in the brain tissues of patients with AD and 5XFAD AD mouse models compared to those of healthy individuals using Western blotting and immunofluorescence staining [44][45][46]. In an in vitro study, the pKr-2 induced neuroinflammation through microglial activation without directly causing neuronal toxicity in the co-culture of mesencephalic neurons and microglia [90]. However, in the hippocampus of the 5XFAD AD mouse model, pKr-2 overexpression was shown to induce microglial activation, leading to excessive neuroinflammation and subsequent neuronal death through the activation of TLR4 transcription factors, such as PU.1 and p-c-Jun [44][45][47][86][89]. Researchers also reported that caffeine and rivaroxaban treatment can suppress the generation and brain influx of pKr-2, leading to a reduction in neuroinflammation and improvement in cognitive impairment in the 5XFAD AD mouse model [46]. Although further detailed mechanistic studies are required to fully understand the microglial activation by pKr-2, various studies have demonstrated that controlling pKr-2 overexpression is a potential therapeutic strategy for AD [44][45][46][47].
2.3. Fibrinogen, Fibrin
Fibrinogen is a key protein involved in the blood-clotting process [34][42][43][54][100][101][102]. The formation of fibrin monomers at the site of vascular injury induces the binding of fibrinogen to the vessel wall and its conversion into fibrin dimers, which form a foam-like structure in the blood and are important for blood clotting [100][101][103]. Recent studies have shown that fibrinogen can traverse the BBB and have significant physiological and pathological effects in the brain [34][42][43][54]. Fibrinogen is associated with neuroinflammation and changes in brain vasculature in neurodegenerative disorders such as AD [42][43]. These changes can cause damage to brain neurons and synapses, leading to symptoms such as cognitive impairment [34]. According to the in vivo study by Merlini et al., fibrinogen can activate microglia in the brain and contribute to the process of removal of synaptic spines, which is associated with the development and progression of AD in mice [34]. Additionally, fibrinogen can impair the integrity of the brain vasculature and cause BBB damage, which occurs in the early stages of AD and increases in severity as the disease progresses [43][102]. Another study suggested that the binding of fibrinogen and Aβ in the brain may exacerbate the severity of a TgCRND8 AD model in vivo [54][55][56], as this association can result in the formation of abnormal blood clots and blockages in the cerebral vasculature [54].
Similar to fibrinogen, fibrin also plays an important role in the process of blood clotting [42][100][101][103][104]. Thrombin cleaves fibrinogen to form fibrin monomers, which polymerize to create the fibrin clot [42][100][101][103][104]. According to a study, conducted in APP/PS1 transgenic mice, an AD model, an increase in fibrin within blood vessels and the abnormal accumulation of the extracellular matrix in the brain tissue were found to induce neurovascular damage and neuroinflammatory responses [104]. The progression of Aβ deposition in the brain tissue was accompanied by vascular damage and inflammatory responses [104], which, in turn, accelerated the progression of AD due to the excessive accumulation of fibrin [104]. Thus, reducing the abnormal accumulation of fibrin in the extracellular matrix and brain vasculature may mitigate brain damage and slow down disease progression [104]. Ryu et al. showed that treatment with the 5B8 antibody targeting the fibrin epitope γ377-395 prevented abnormal fibrin accumulation in a 5XFAD AD animal model, resulting in decreased neuroinflammation and neuronal damage [42].
2.4. Other Proteins and Factors
In addition to coagulation factors, blood vessels contain a diverse array of proteins and factors that serve a range of functions beyond their role in hemostasis [68][105][106][107][108][109][110][111][112][113][114]. These proteins and factors can also infiltrate the brain and have harmful effects [108][109][110][111][112][113][114]. In one study, the blood-borne Aβ protein was found to penetrate the BBB and accumulate in the microglia, leading to AD pathogenesis [114]. The blood Aβ protein induces neuroinflammation and microglial activation, leading to the formation of brain deposits and neuronal damage [114]. AD patients have increased levels of albumin and immunoglobulin G (IgG) in their blood compared to normal individuals, and the passage of these proteins through the impaired BBB into the brain may play a role in the pathophysiology of AD [108][110][113]. Additionally, the brain tissues of patients with AD patients were shown to have increased concentration of hemoglobin-derived peptides, which may contribute to neuroinflammation and neuronal damage [112]. Another study reported increased iron levels in the hippocampal region of patients with AD using magnetic resonance imaging, which may be related to the pathophysiological changes in AD [111]. Plasminogen derived from blood was shown to induce Aβ accumulation in the brain through microglial activation, further aggravating the neuroinflammatory responses [109]. Specifically, plasminogens derived from blood could induce Aβ accumulation in the brain through microglial activation, leading to further promotion of neuroinflammatory responses [109]. Therefore, further investigation is required to examine additional proteins and factors present in the blood and to investigate their mechanisms of infiltration into the brain and activation of microglial cells.
In addition, the overexpression or overactivation of certain brain proteins could disrupt BBB integrity. A study investigated the role of IL-1β in regulating BBB permeability and found that IL-1β activated microglia, leading to damage and inflammation of vascular endothelial cells; this, subsequently, affected angiogenesis and increased vascular permeability [115]. In addition, an investigation of BBB destruction and cell death in an ischemic stroke model revealed that the TNF-α derived from microglia induced neuronal death, exacerbating BBB destruction [116]. Some studies revealed that blood-derived extracellular vesicles, such as microvesicles, transported into the brain could influence microglial activation, and extracellular vesicles generated through microglial activation could induce neuronal damage in the AD brain [117][118]. Moreover, High Mobility Group Box-1 (HMGB1) was identified as one of the key factors that promotes BBB breakdown. HMGB1 increases in the setting of brain injury or inflammation, inducing an inflammatory response in the endothelial cells of the BBB and increasing BBB permeability [119]. Matrix metalloproteinases (MMPs) are protein-degrading enzymes that regulate the extracellular matrix, but excessive MMP activity can cause brain tissue damage and inflammation [119][120]. Increased activity of MMP-9 was shown to be involved in the breakdown of the BBB [120]. Therefore, further research on the signaling pathways such as MMP-9 and other proteins in relation to BBB damage may support the development of new therapeutic strategies for AD.
References
- Cummings, J.L. Alzheimer’s disease. N. Engl. J. Med. 2004, 351, 56–67.
- Burns, A.; Iliffe, S. Alzheimer’s disease. BMJ 2009, 338, b158.
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32.
- Bondi, M.W.; Edmonds, E.C.; Salmon, D.P. Alzheimer’s Disease: Past, Present, and Future. J. Int. Neuropsychol. Soc. 2017, 23, 818–831.
- Brayne, C.; Miller, B. Dementia and aging populations-A global priority for contextualized research and health policy. PLoS Med. 2017, 14, e1002275.
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581.
- Adler, D.H.; Wisse, L.E.M.; Ittyerah, R.; Pluta, J.B.; Ding, S.L.; Xie, L.; Wang, J.; Kadivar, S.; Robinson, J.L.; Schuck, T.; et al. Characterizing the human hippocampus in aging and Alzheimer’s disease using a computational atlas derived from ex vivo MRI and histology. Proc. Natl. Acad. Sci. USA 2018, 115, 4252–4257.
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344.
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227.
- Tanzi, R.E. The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006296.
- Cuyvers, E.; Sleegers, K. Genetic variations underlying Alzheimer’s disease: Evidence from genome-wide association studies and beyond. Lancet Neurol. 2016, 15, 857–868.
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430.
- Krause, D.L.; Muller, N. Neuroinflammation, microglia and implications for anti-inflammatory treatment in Alzheimer’s disease. Int. J. Alzheimer’s Dis. 2010, 2010, 732806.
- Mandrekar-Colucci, S.; Landreth, G.E. Microglia and inflammation in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 156–167.
- Perry, V.H.; Nicoll, J.A.; Holmes, C. Microglia in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 193–201.
- Perry, V.H.; Holmes, C. Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 2014, 10, 217–224.
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136.
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249.
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472.
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369.
- Hensley, K. Neuroinflammation in Alzheimer’s disease: Mechanisms, pathologic consequences, and potential for therapeutic manipulation. J. Alzheimer’s Dis. 2010, 21, 1–14.
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405.
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372.
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. 2018, 4, 575–590.
- Wilcock, D.M. A changing perspective on the role of neuroinflammation in Alzheimer’s disease. Int. J. Alzheimer’s Dis. 2012, 2012, 495243.
- Alibhai, J.D.; Diack, A.B.; Manson, J.C. Unravelling the glial response in the pathogenesis of Alzheimer’s disease. FASEB J. 2018, 32, 5766–5777.
- Prokop, S.; Miller, K.R.; Heppner, F.L. Microglia actions in Alzheimer’s disease. Acta Neuropathol. 2013, 126, 461–477.
- Lee, C.Y.; Landreth, G.E. The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 2010, 117, 949–960.
- Katsumoto, A.; Takeuchi, H.; Takahashi, K.; Tanaka, F. Microglia in Alzheimer’s Disease: Risk Factors and Inflammation. Front. Neurol. 2018, 9, 978.
- McCaulley, M.E.; Grush, K.A. Alzheimer’s Disease: Exploring the Role of Inflammation and Implications for Treatment. Int. J. Alzheimer’s Dis. 2015, 2015, 515248.
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25.
- Abbott, N.J. Dynamics of CNS barriers: Evolution, differentiation, and modulation. Cell. Mol. Neurobiol. 2005, 25, 5–23.
- Lee, J.M.; Kim, S.R. Prothrombin kringle-2, a mediator of microglial activation: New insight in Alzheimer’s disease pathogenesis. Neural Regen. Res. 2022, 17, 2675–2676.
- Merlini, M.; Rafalski, V.A.; Rios Coronado, P.E.; Gill, T.M.; Ellisman, M.; Muthukumar, G.; Subramanian, K.S.; Ryu, J.K.; Syme, C.A.; Davalos, D.; et al. Fibrinogen Induces Microglia-Mediated Spine Elimination and Cognitive Impairment in an Alzheimer’s Disease Model. Neuron 2019, 101, 1099–1108.e1096.
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276.
- van de Haar, H.J.; Burgmans, S.; Jansen, J.F.; van Osch, M.J.; van Buchem, M.A.; Muller, M.; Hofman, P.A.; Verhey, F.R.; Backes, W.H. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 2016, 281, 527–535.
- Ujiie, M.; Dickstein, D.L.; Carlow, D.A.; Jefferies, W.A. Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation 2003, 10, 463–470.
- Kawas, C.H. Clinical practice. Early Alzheimer’s disease. N. Engl. J. Med. 2003, 349, 1056–1063.
- Henneman, W.J.; Sluimer, J.D.; Barnes, J.; van der Flier, W.M.; Sluimer, I.C.; Fox, N.C.; Scheltens, P.; Vrenken, H.; Barkhof, F. Hippocampal atrophy rates in Alzheimer disease: Added value over whole brain volume measures. Neurology 2009, 72, 999–1007.
- Xiao, M.; Xiao, Z.J.; Yang, B.; Lan, Z.; Fang, F. Blood-Brain Barrier: More Contributor to Disruption of Central Nervous System Homeostasis Than Victim in Neurological Disorders. Front. Neurosci. 2020, 14, 764.
- Desai, B.S.; Monahan, A.J.; Carvey, P.M.; Hendey, B. Blood-brain barrier pathology in Alzheimer’s and Parkinson’s disease: Implications for drug therapy. Cell Transplant. 2007, 16, 285–299.
- Ryu, J.K.; Rafalski, V.A.; Meyer-Franke, A.; Adams, R.A.; Poda, S.B.; Rios Coronado, P.E.; Pedersen, L.O.; Menon, V.; Baeten, K.M.; Sikorski, S.L.; et al. Fibrin-targeting immunotherapy protects against neuroinflammation and neurodegeneration. Nat. Immunol. 2018, 19, 1212–1223.
- Ryu, J.K.; McLarnon, J.G. A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. J. Cell. Mol. Med. 2009, 13, 2911–2925.
- Shin, W.H.; Jeon, M.T.; Leem, E.; Won, S.Y.; Jeong, K.H.; Park, S.J.; McLean, C.; Lee, S.J.; Jin, B.K.; Jung, U.J.; et al. Induction of microglial toll-like receptor 4 by prothrombin kringle-2: A potential pathogenic mechanism in Parkinson’s disease. Sci. Rep. 2015, 5, 14764.
- Leem, E.; Jeong, K.H.; Won, S.Y.; Shin, W.H.; Kim, S.R. Prothrombin Kringle-2: A Potential Inflammatory Pathogen in the Parkinsonian Dopaminergic System. Exp. Neurobiol. 2016, 25, 147–155.
- Kim, S.; Moon, G.J.; Kim, H.J.; Kim, D.G.; Kim, J.; Nam, Y.; Sharma, C.; Leem, E.; Lee, S.; Kim, K.S.; et al. Control of hippocampal prothrombin kringle-2 (pKr-2) expression reduces neurotoxic symptoms in five familial Alzheimer’s disease mice. Br. J. Pharmacol. 2022, 179, 998–1016.
- Kim, S.; Sharma, C.; Shin, M.; Kim, H.J.; Kim, J.; Kim, S.R. pKr-2 induces neurodegeneration via upregulation of microglial TLR4 in the hippocampus of AD brain. Brain Behav. Immun. Health 2023, 28, 100593.
- Arai, T.; Miklossy, J.; Klegeris, A.; Guo, J.P.; McGeer, P.L. Thrombin and prothrombin are expressed by neurons and glial cells and accumulate in neurofibrillary tangles in Alzheimer disease brain. J. Neuropathol. Exp. Neurol. 2006, 65, 19–25.
- Choi, S.H.; Lee, D.Y.; Kim, S.U.; Jin, B.K. Thrombin-induced oxidative stress contributes to the death of hippocampal neurons in vivo: Role of microglial NADPH oxidase. J. Neurosci. 2005, 25, 4082–4090.
- Grammas, P.; Martinez, J.M. Targeting thrombin: An inflammatory neurotoxin in Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 42 (Suppl. S4), S537–S544.
- Iannucci, J.; Renehan, W.; Grammas, P. Thrombin, a Mediator of Coagulation, Inflammation, and Neurotoxicity at the Neurovascular Interface: Implications for Alzheimer’s Disease. Front. Neurosci. 2020, 14, 762.
- Brewer, G.J. Thrombin causes cell spreading and redistribution of beta-amyloid immunoreactivity in cultured hippocampal neurons. J. Neurochem. 1996, 67, 119–130.
- Ciallella, J.R.; Figueiredo, H.; Smith-Swintosky, V.; McGillis, J.P. Thrombin induces surface and intracellular secretion of amyloid precursor protein from human endothelial cells. Thromb. Haemost. 1999, 81, 630–637.
- Cortes-Canteli, M.; Paul, J.; Norris, E.H.; Bronstein, R.; Ahn, H.J.; Zamolodchikov, D.; Bhuvanendran, S.; Fenz, K.M.; Strickland, S. Fibrinogen and beta-amyloid association alters thrombosis and fibrinolysis: A possible contributing factor to Alzheimer’s disease. Neuron 2010, 66, 695–709.
- Fan, D.Y.; Sun, H.L.; Sun, P.Y.; Jian, J.M.; Li, W.W.; Shen, Y.Y.; Zeng, F.; Wang, Y.J.; Bu, X.L. The Correlations Between Plasma Fibrinogen With Amyloid-Beta and Tau Levels in Patients With Alzheimer’s Disease. Front. Neurosci. 2020, 14, 625844.
- Page, M.J.; Thomson, G.J.A.; Nunes, J.M.; Engelbrecht, A.M.; Nell, T.A.; de Villiers, W.J.S.; de Beer, M.C.; Engelbrecht, L.; Kell, D.B.; Pretorius, E. Serum amyloid A binds to fibrin(ogen), promoting fibrin amyloid formation. Sci. Rep. 2019, 9, 3102.
- Sulimai, N.; Brown, J.; Lominadze, D. The Effects of Fibrinogen’s Interactions with Its Neuronal Receptors, Intercellular Adhesion Molecule-1 and Cellular Prion Protein. Biomolecules 2021, 11, 1381.
- Michalicova, A.; Majerova, P.; Kovac, A. Tau Protein and Its Role in Blood-Brain Barrier Dysfunction. Front. Mol. Neurosci. 2020, 13, 570045.
- Iqbal, K.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I. Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664.
- Ronaldson, P.T.; Davis, T.P. Regulation of blood-brain barrier integrity by microglia in health and disease: A therapeutic opportunity. J. Cereb. Blood Flow Metab. 2020, 40, S6–S24.
- Dyrna, F.; Hanske, S.; Krueger, M.; Bechmann, I. The blood-brain barrier. J. Neuroimmune Pharmacol. 2013, 8, 763–773.
- Sonar, S.A.; Lal, G. Blood-brain barrier and its function during inflammation and autoimmunity. J. Leukoc. Biol. 2018, 103, 839–853.
- Reed, M.J.; Damodarasamy, M.; Banks, W.A. The extracellular matrix of the blood-brain barrier: Structural and functional roles in health, aging, and Alzheimer’s disease. Tissue Barriers 2019, 7, 1651157.
- Verheggen, I.C.M.; de Jong, J.J.A.; van Boxtel, M.P.J.; Postma, A.A.; Jansen, J.F.A.; Verhey, F.R.J.; Backes, W.H. Imaging the role of blood-brain barrier disruption in normal cognitive ageing. Geroscience 2020, 42, 1751–1764.
- Erdo, F.; Denes, L.; de Lange, E. Age-associated physiological and pathological changes at the blood-brain barrier: A review. J. Cereb. Blood Flow Metab. 2017, 37, 4–24.
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302.
- Goodall, E.F.; Wang, C.; Simpson, J.E.; Baker, D.J.; Drew, D.R.; Heath, P.R.; Saffrey, M.J.; Romero, I.A.; Wharton, S.B. Age-associated changes in the blood-brain barrier: Comparative studies in human and mouse. Neuropathol. Appl. Neurobiol. 2018, 44, 328–340.
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078.
- Grossmann, K. Anticoagulants for Treatment of Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 77, 1373–1382.
- Mann, K.G. Prothrombin. Methods Enzymol. 1976, 45, 123–156.
- Samama, C.M. Prothrombin complex concentrates: A brief review. Eur. J. Anaesthesiol. 2008, 25, 784–789.
- Davie, E.W.; Fujikawa, K.; Kisiel, W. The coagulation cascade: Initiation, maintenance, and regulation. Biochemistry 1991, 30, 10363–10370.
- Stojanovski, B.M.; Di Cera, E. Role of sequence and position of the cleavage sites in prothrombin activation. J. Biol. Chem. 2021, 297, 100955.
- Sokolova, E.; Reiser, G. Prothrombin/thrombin and the thrombin receptors PAR-1 and PAR-4 in the brain: Localization, expression and participation in neurodegenerative diseases. Thromb. Haemost. 2008, 100, 576–581.
- Krenzlin, H.; Lorenz, V.; Danckwardt, S.; Kempski, O.; Alessandri, B. The Importance of Thrombin in Cerebral Injury and Disease. Int. J. Mol. Sci. 2016, 17, 84.
- Ishida, Y.; Nagai, A.; Kobayashi, S.; Kim, S.U. Upregulation of protease-activated receptor-1 in astrocytes in Parkinson disease: Astrocyte-mediated neuroprotection through increased levels of glutathione peroxidase. J. Neuropathol. Exp. Neurol. 2006, 65, 66–77.
- Davie, E.W.; Kulman, J.D. An overview of the structure and function of thrombin. Semin. Thromb. Hemost. 2006, 32 (Suppl. S1), 3–15.
- Al-Amer, O.M. The role of thrombin in haemostasis. Blood Coagul. Fibrinolysis 2022, 33, 145–148.
- Suo, Z.; Citron, B.A.; Festoff, B.W. Thrombin: A potential proinflammatory mediator in neurotrauma and neurodegenerative disorders. Curr. Drug Targets Inflamm. Allergy 2004, 3, 105–114.
- Suo, Z.; Wu, M.; Ameenuddin, S.; Anderson, H.E.; Zoloty, J.E.; Citron, B.A.; Andrade-Gordon, P.; Festoff, B.W. Participation of protease-activated receptor-1 in thrombin-induced microglial activation. J. Neurochem. 2002, 80, 655–666.
- Yin, M.; Chen, Z.; Ouyang, Y.; Zhang, H.; Wan, Z.; Wang, H.; Wu, W.; Yin, X. Thrombin-induced, TNFR-dependent miR-181c downregulation promotes MLL1 and NF-kappaB target gene expression in human microglia. J. Neuroinflam. 2017, 14, 132.
- Han, C.; Xia, X.; Jiao, S.; Li, G.; Ran, Q.; Yao, S. Tripartite motif containing protein 37 involves in thrombin stimulated BV-2 microglial cell apoptosis and interleukin 1beta release. Biochem. Biophys. Res. Commun. 2019, 516, 1252–1257.
- Suo, Z.; Wu, M.; Citron, B.A.; Palazzo, R.E.; Festoff, B.W. Rapid tau aggregation and delayed hippocampal neuronal death induced by persistent thrombin signaling. J. Biol. Chem. 2003, 278, 37681–37689.
- Park, S.Y.; Ferreira, A. The generation of a 17 kDa neurotoxic fragment: An alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J. Neurosci. 2005, 25, 5365–5375.
- Garg, S.; Timm, T.; Mandelkow, E.M.; Mandelkow, E.; Wang, Y. Cleavage of Tau by calpain in Alzheimer’s disease: The quest for the toxic 17 kD fragment. Neurobiol. Aging 2011, 32, 1–14.
- Chung, Y.C.; Jeong, J.Y.; Jin, B.K. Interleukin-4-Mediated Oxidative Stress Is Harmful to Hippocampal Neurons of Prothrombin Kringle-2-Lesioned Rat In Vivo. Antioxidants 2020, 9, 1068.
- Dasgupta, S.K.; Thiagarajan, P. Inhibition of thrombin activity by prothrombin activation fragment 1.2. J. Thromb. Thrombolysis 2007, 24, 157–162.
- Jeong, J.Y.; Wi, R.; Chung, Y.C.; Jin, B.K. Interleukin-13 Propagates Prothrombin Kringle-2-Induced Neurotoxicity in Hippocampi In Vivo via Oxidative Stress. Int. J. Mol. Sci. 2021, 22, 3486.
- Kim, S.; Moon, G.J.; Oh, Y.S.; Park, J.; Shin, W.H.; Jeong, J.Y.; Choi, K.S.; Jin, B.K.; Kholodilov, N.; Burke, R.E.; et al. Protection of nigral dopaminergic neurons by AAV1 transduction with Rheb(S16H) against neurotoxic inflammation in vivo. Exp. Mol. Med. 2018, 50, e440.
- Kim, S.R.; Chung, E.S.; Bok, E.; Baik, H.H.; Chung, Y.C.; Won, S.Y.; Joe, E.; Kim, T.H.; Kim, S.S.; Jin, M.Y.; et al. Prothrombin kringle-2 induces death of mesencephalic dopaminergic neurons in vivo and in vitro via microglial activation. J. Neurosci. Res 2010, 88, 1537–1548.
- Kim, T.H.; Oh, S.; Kim, S.S. Recombinant human prothrombin kringle-2 induces bovine capillary endothelial cell cycle arrest at G0-G1 phase through inhibition of cyclin D1/CDK4 complex: Modulation of reactive oxygen species generation and up-regulation of cyclin-dependent kinase inhibitors. Angiogenesis 2005, 8, 307–314.
- Lee, T.H.; Rhim, T.; Kim, S.S. Prothrombin kringle-2 domain has a growth inhibitory activity against basic fibroblast growth factor-stimulated capillary endothelial cells. J. Biol. Chem. 1998, 273, 28805–28812.
- Leem, E.; Oh, Y.S.; Shin, W.H.; Jin, B.K.; Jeong, J.Y.; Shin, M.; Kim, D.W.; Jang, J.H.; Kim, H.J.; Ha, C.M.; et al. Effects of Silibinin Against Prothrombin Kringle-2-Induced Neurotoxicity in the Nigrostriatal Dopaminergic System In Vivo. J. Med. Food 2019, 22, 277–285.
- Nam, J.H.; Leem, E.; Jeon, M.T.; Kim, Y.J.; Jung, U.J.; Choi, M.S.; Maeng, S.; Jin, B.K.; Kim, S.R. Inhibition of prothrombin kringle-2-induced inflammation by minocycline protects dopaminergic neurons in the substantia nigra in vivo. Neuroreport 2014, 25, 489–495.
- Rhim, T.Y.; Park, C.S.; Kim, E.; Kim, S.S. Human prothrombin fragment 1 and 2 inhibit bFGF-induced BCE cell growth. Biochem. Biophys. Res. Commun. 1998, 252, 513–516.
- Ryu, J.; Min, K.J.; Rhim, T.Y.; Kim, T.H.; Pyo, H.; Jin, B.; Kim, S.U.; Jou, I.; Kim, S.S.; Joe, E.H. Prothrombin kringle-2 activates cultured rat brain microglia. J. Immunol. 2002, 168, 5805–5810.
- Taneda, H.; Andoh, K.; Nishioka, J.; Takeya, H.; Suzuki, K. Blood coagulation factor Xa interacts with a linear sequence of the kringle 2 domain of prothrombin. J. Biochem. 1994, 116, 589–597.
- Won, S.Y.; Choi, S.H.; Jin, B.K. Prothrombin kringle-2-induced oxidative stress contributes to the death of cortical neurons in vivo and in vitro: Role of microglial NADPH oxidase. J. Neuroimmunol. 2009, 214, 83–92.
- Deguchi, H.; Takeya, H.; Gabazza, E.C.; Nishioka, J.; Suzuki, K. Prothrombin kringle 1 domain interacts with factor Va during the assembly of prothrombinase complex. Biochem. J. 1997, 321 Pt 3, 729–735.
- Litvinov, R.I.; Pieters, M.; de Lange-Loots, Z.; Weisel, J.W. Fibrinogen and Fibrin. Subcell. Biochem. 2021, 96, 471–501.
- Pieters, M.; Wolberg, A.S. Fibrinogen and fibrin: An illustrated review. Res. Pract. Thromb. Haemost. 2019, 3, 161–172.
- Davalos, D.; Ryu, J.K.; Merlini, M.; Baeten, K.M.; Le Moan, N.; Petersen, M.A.; Deerinck, T.J.; Smirnoff, D.S.; Bedard, C.; Hakozaki, H.; et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat. Commun. 2012, 3, 1227.
- Weisel, J.W.; Litvinov, R.I. Fibrin Formation, Structure and Properties. Subcell. Biochem. 2017, 82, 405–456.
- Paul, J.; Strickland, S.; Melchor, J.P. Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer’s disease. J. Exp. Med. 2007, 204, 1999–2008.
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596.
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201.
- Engelhardt, B.; Sorokin, L. The blood-brain and the blood-cerebrospinal fluid barriers: Function and dysfunction. Semin. Immunopathol. 2009, 31, 497–511.
- Alafuzoff, I.; Adolfsson, R.; Bucht, G.; Winblad, B. Albumin and immunoglobulin in plasma and cerebrospinal fluid, and blood-cerebrospinal fluid barrier function in patients with dementia of Alzheimer type and multi-infarct dementia. J. Neurol. Sci. 1983, 60, 465–472.
- Baker, S.K.; Chen, Z.L.; Norris, E.H.; Revenko, A.S.; MacLeod, A.R.; Strickland, S. Blood-derived plasminogen drives brain inflammation and plaque deposition in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E9687–E9696.
- Hampel, H.; Kotter, H.U.; Moller, H.J. Blood-cerebrospinal fluid barrier dysfunction for high molecular weight proteins in Alzheimer disease and major depression: Indication for disease subsets. Alzheimer Dis. Assoc. Disord. 1997, 11, 78–87.
- Raven, E.P.; Lu, P.H.; Tishler, T.A.; Heydari, P.; Bartzokis, G. Increased iron levels and decreased tissue integrity in hippocampus of Alzheimer’s disease detected in vivo with magnetic resonance imaging. J. Alzheimer’s Dis. 2013, 37, 127–136.
- Slemmon, J.R.; Hughes, C.M.; Campbell, G.A.; Flood, D.G. Increased levels of hemoglobin-derived and other peptides in Alzheimer’s disease cerebellum. J. Neurosci. 1994, 14, 2225–2235.
- Tibbling, G.; Link, H.; Ohman, S. Principles of albumin and IgG analyses in neurological disorders. I. Establishment of reference values. Scand. J. Clin. Lab. Investig. 1977, 37, 385–390.
- Bu, X.L.; Xiang, Y.; Jin, W.S.; Wang, J.; Shen, L.L.; Huang, Z.L.; Zhang, K.; Liu, Y.H.; Zeng, F.; Liu, J.H.; et al. Blood-derived amyloid-beta protein induces Alzheimer’s disease pathologies. Mol. Psychiatry 2018, 23, 1948–1956.
- Argaw, A.T.; Zhang, Y.; Snyder, B.J.; Zhao, M.L.; Kopp, N.; Lee, S.C.; Raine, C.S.; Brosnan, C.F.; John, G.R. IL-1beta regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. J. Immunol. 2006, 177, 5574–5584.
- Chen, A.Q.; Fang, Z.; Chen, X.L.; Yang, S.; Zhou, Y.F.; Mao, L.; Xia, Y.P.; Jin, H.J.; Li, Y.N.; You, M.F.; et al. Microglia-derived TNF-alpha mediates endothelial necroptosis aggravating blood brain-barrier disruption after ischemic stroke. Cell Death Dis. 2019, 10, 487.
- Trotta, T.; Panaro, M.A.; Cianciulli, A.; Mori, G.; Di Benedetto, A.; Porro, C. Microglia-derived extracellular vesicles in Alzheimer’s Disease: A double-edged sword. Biochem. Pharmacol. 2018, 148, 184–192.
- Marostica, G.; Gelibter, S.; Gironi, M.; Nigro, A.; Furlan, R. Extracellular Vesicles in Neuroinflammation. Front. Cell Dev. Biol. 2020, 8, 623039.
- Nishibori, M.; Wang, D.; Ousaka, D.; Wake, H. High Mobility Group Box-1 and Blood-Brain Barrier Disruption. Cells 2020, 9, 2650.
- Brilha, S.; Ong, C.W.M.; Weksler, B.; Romero, N.; Couraud, P.O.; Friedland, J.S. Matrix metalloproteinase-9 activity and a downregulated Hedgehog pathway impair blood-brain barrier function in an in vitro model of CNS tuberculosis. Sci. Rep. 2017, 7, 16031.
More
Information
Subjects:
Immunology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
810
Revisions:
2 times
(View History)
Update Date:
17 May 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No