Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Immunology
The blood–brain barrier (BBB) restricts entry of neurotoxic plasma components, blood cells, and pathogens into the brain, leading to proper neuronal functioning. BBB impairment leads to blood-borne protein infiltration such as prothrombin, thrombin, prothrombin kringle-2, fibrinogen, fibrin, and other harmful substances. Thus, microglial activation and release of pro-inflammatory mediators commence, resulting in neuronal damage and leading to impaired cognition via neuroinflammatory responses, which are important features observed in the brain of Alzheimer’s disease (AD) patients.
- Alzheimer’s disease
- microglia
- neuroinflammation
- neurodegeneration
- blood–brain barrier
1. Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder often characterized by memory loss and impaired cognition [1,2,3,4]. The global AD prevalence is increasing concurrently with the aging population [4,5,6,7]. However, the pathogenic mechanisms of AD remain elusive, despite extensive research on the various causative and contributory factors, including genetics [1,2,3,8,9,10,11,12]. An accumulating body of evidence suggests that neuroinflammation mediated by glial activation is an important neurotoxic mechanism associated with the initiation and progression of AD, including the subsequent neuronal cell death and cognitive impairment [13,14,15,16,17,18,19,20,21,22,23,24].
Microglia promote brain homeostasis through synaptic remodeling maintenance and dead cell removal under non-pathological conditions. However, during the early stages of AD, an increase in inflammatory responses via microglial activation in the brain has been reported [23,25,26]. These responses are also involved in the early formation of amyloid beta (Aβ) plaques and persistent microglial activation, which can result in the generation of pro-inflammatory cytokines and reactive oxygen species (ROS) [23,25,26]. These pathways interact and reinforce one another to induce pathological alterations in the AD brain, which primarily involve microglial activation, neuroinflammation, tau hyperphosphorylation, compromised mitochondrial activity, and oxidative stress [23,25,26]. This, in turn, triggers neurodegeneration due to the subsequent loss of synapses and neurons, resulting in neuronal death [14,15,17,18,20]. In particular, the initial microglial activation and the resulting neuroinflammation may act as a starting point for the observed neurodegeneration in the AD brain [23,25,26]. These findings suggest that initial microglial activation can exacerbate AD progression, despite potential variations in the roles of microglia activation and inflammatory responses within the brain. [13,17,18,27,28,29,30]. Therefore, there is a pressing need to investigate such endogenous substances that may induce microglial activation to build an effective AD treatment strategy.
The blood–brain barrier (BBB) is essential for the maintenance of the brain tissue microenvironment. It improves connectivity while also providing insulation of the brain parenchyma from the peripheral circulation system [31]. It is composed of specialized endothelial cells and various tight junction (TJ) proteins to ensure optimal functioning of the BBB [31]. These TJs, together with specialized transporters, allow the passage of nutrients into the central nervous system (CNS) while restricting blood-borne molecule influx [32]. Meanwhile, in pathological conditions, BBB disruption can result in the entry of blood-borne proteins such as prothrombin, thrombin, prothrombin kringle-2 (pKr-2), fibrinogen, and fibrin, among others, into the brain parenchyma [33,34]. This causes gradual neurodegeneration and neuronal death through direct and/or indirect neurotoxicity, oxidative stress, mitochondrial function loss, and pro-inflammatory mediator release, leading to memory loss and a decline in cognitive function. Several reports suggest that the early stages of AD are critical, as BBB breakdown occurs during this period [35,36,37,38,39], which allows the entry of blood-borne proteins in the brain parenchyma, thus contributing to neurotoxicity by several mechanisms [40,41]. For instance, blood-borne proteins such as prothrombin, thrombin, pKr-2, fibrinogen, and fibrin can activate microglia and induce neuroinflammation, resulting in neurodegeneration and cognitive decline in various animal models of AD [34,42,43,44,45,46,47,48,49,50,51]. Moreover, some of these blood proteins (such as thrombin, fibrinogen, and fibrin) can cluster with Aβ, the main causal agent of AD, and can further exacerbate microglial activation [52,53,54,55,56]. Furthermore, fibrinogen deposition can lead to impaired memory and result in exacerbated levels of interleukin-6, ROS, mitochondrial superoxide, and nitrite in mouse brain neurons [57]. This can result in abnormal tau phosphorylation, which is another major hallmark of AD [58], and further amplify neurotoxicity through the interplay with Aβ plaques, thus affecting neurotransmitter balance [59]. As fibrinogen is not expressed in the brain, BBB disruption is required for plasma fibrinogen to cross the BBB and reach the brain parenchyma [34]. Therefore, investigating the mechanisms of blood-borne protein entry into the brain and microglial activation promotion leading to the release of pro-inflammatory cytokines resulting in neuronal loss may help develop novel therapeutic strategies for AD.
2. Blood-Borne Protein by BBB Leakage Induces Microglial Activation in AD
The BBB prevents the entry of harmful substances into the brain from the blood [72,88,108,109]. Age-related degeneration of the BBB increases its permeability along with a decrease in cerebral blood flow [95,109,110,111,112]. A decrease in the interaction between neurons and endothelial cells and astrocytes is another factor that contributes to the BBB breakdown and impairment. This results in the blood-borne substance leakage into the brain, including prothrombin, thrombin, pKr-2, fibrinogen, fibrin, and albumin (Figure 1) [34,46,51,86].
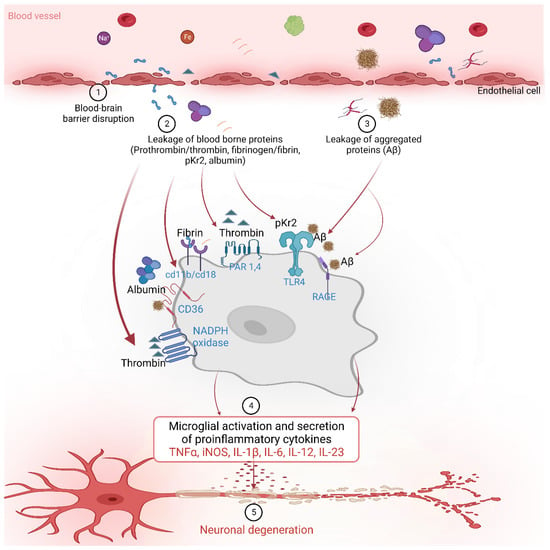
Figure 1. The blood-borne proteins interact with various receptors on the microglial cells to activate downstream signaling and influence various inflammatory and neurodegenerative processes in Alzheimer’s disease.
2.1. Prothrombin and Thrombin
Prothrombin is a protein involved in the blood coagulation pathway, which helps to form blood clots in response to injury [113,114,115,116,117]. Prothrombin in blood plasma is converted into thrombin by factor X or prothrombinase, which then transforms fibrinogen into fibrin to form a clot with platelets, preventing blood loss [113,114,115,116,117]. Increased expression of prothrombin has been found in both neurons and glial cells in the brain tissue of patients with AD patients compared with healthy controls [48,50,118,119,120]. Furthermore, prothrombin has been shown to accumulate in neurofibrillary tangles, a pathological hallmark of AD [48]. Although the precise mechanisms underlying the upregulation of prothrombin and thrombin in AD are not completely understood, a growing body of evidence suggests their potential role in the pathogenesis of this neurodegenerative disease [48,49,50,51,118,119]. These findings suggest that prothrombin may contribute to the neurodegenerative process in AD, particularly concerning neuronal damage [48,118,120]. Thrombin is a protease that is also involved in the blood coagulation pathway, but it can also be generated in the brain in response to injury and inflammation [121,122,123]. In AD, thrombin is upregulated in the brain and may contribute to the pathogenesis of the disease [48,50,118]. In one study, the expression of prothrombin mRNA in brain tissues, neuroblastoma cells, cultured human astrocytes, oligodendrocytes, and microglial cells were detected using the reverse transcriptase–polymerase chain reaction, and the presence of prothrombin and thrombin proteins in the brain was confirmed using specific monoclonal and polyclonal antibodies for both proteins through immunohistochemistry [48]. Furthermore, studies have demonstrated increased expression of thrombin and its receptors such as protease-activated receptor 1 (PAR-1) and protease-activated receptor 4 (PAR-4) in the brain in neurodegenerative diseases [118,124]. Thrombin has also been reported to induce the NADPH oxidase in microglia in the hippocampus, leading to the death of neuronal cells, suggesting a potential mechanism by which thrombin contributes to neurodegenerative processes in the brain [49]. Other reports also show that thrombin can activate microglia, leading to pro-inflammatory cytokine releases such as TNF-α and IL-1β in BV-2 and primary human microglia in vitro [125,126].
Thrombin-induced neuroinflammation via microglial activation was found to be mediated through the PAR-1 receptor signaling pathway [124]. In the AD brain, thrombin can also induce vascular inflammation, exacerbating BBB damage [50,51]. Thrombin may also contribute to Aβ accumulation in the brain, as it can cleave the Aβ precursor protein and generate Aβ peptides that aggregate and form amyloid plaques in the cultured hippocampal neurons and human endothelial cells in vitro [52,53]. Additionally, reports suggest that thrombin can induce tau hyperphosphorylation and aggregation in murine hippocampal neurons, resulting in microglial activation, reduction in delayed synaptophysin, and apoptotic neuronal death [127]. Moreover, along with calpain-1, it can cleave tau, resulting in a ~17 kDA neurotoxic fragment that can cause impaired synaptic function and axonal transport, mitochondrial membrane depolarization, and behavioral deficits in AD brains [128,129].
2.2. pKr-2
pKr-2, also known as fragment 2, is a small protein fragment that is released during the activation of prothrombin [44,45,46,47,130,131,132,133,134,135,136,137,138,139,140,141,142], which is a precursor protein that is converted into the active blood-clotting enzyme thrombin [44,45,46,47,130,131,132,133,134,135,136,137,138,139,140,141,142]. The role of pKr-2 in the blood is to inhibit the prothrombinase complex activity in conjunction with Factor Va, thereby preventing further thrombin production [45,139,143]. In addition, pKr-2 inhibits endothelial cell proliferation and angiogenesis, suggesting its potential role in BBB maintenance [135,136].
We recently reported the increased pKr-2 protein expression in the brain tissues of patients with AD and 5XFAD AD mouse models compared to those of healthy individuals using Western blotting and immunofluorescence staining [44,45,46]. In an in vitro study, the pKr-2 induced neuroinflammation through microglial activation without directly causing neuronal toxicity in the co-culture of mesencephalic neurons and microglia [134]. However, in the hippocampus of the 5XFAD AD mouse model, pKr-2 overexpression was shown to induce microglial activation, leading to excessive neuroinflammation and subsequent neuronal death through the activation of TLR4 transcription factors, such as PU.1 and p-c-Jun [44,45,47,130,133]. We also reported that caffeine and rivaroxaban treatment can suppress the generation and brain influx of pKr-2, leading to a reduction in neuroinflammation and improvement in cognitive impairment in the 5XFAD AD mouse model [46]. Although further detailed mechanistic studies are required to fully understand the microglial activation by pKr-2, various studies have demonstrated that controlling pKr-2 overexpression is a potential therapeutic strategy for AD [44,45,46,47].
2.3. Fibrinogen, Fibrin
Fibrinogen is a key protein involved in the blood-clotting process [34,42,43,54,144,145,146]. The formation of fibrin monomers at the site of vascular injury induces the binding of fibrinogen to the vessel wall and its conversion into fibrin dimers, which form a foam-like structure in the blood and are important for blood clotting [144,145,147]. Recent studies have shown that fibrinogen can traverse the BBB and have significant physiological and pathological effects in the brain [34,42,43,54]. Fibrinogen is associated with neuroinflammation and changes in brain vasculature in neurodegenerative disorders such as AD [42,43]. These changes can cause damage to brain neurons and synapses, leading to symptoms such as cognitive impairment [34]. According to the in vivo study by Merlini et al., fibrinogen can activate microglia in the brain and contribute to the process of removal of synaptic spines, which is associated with the development and progression of AD in mice [34]. Additionally, fibrinogen can impair the integrity of the brain vasculature and cause BBB damage, which occurs in the early stages of AD and increases in severity as the disease progresses [43,146]. Another study suggested that the binding of fibrinogen and Aβ in the brain may exacerbate the severity of a TgCRND8 AD model in vivo [54,55,56], as this association can result in the formation of abnormal blood clots and blockages in the cerebral vasculature [54].
Similar to fibrinogen, fibrin also plays an important role in the process of blood clotting [42,144,145,147,148]. Thrombin cleaves fibrinogen to form fibrin monomers, which polymerize to create the fibrin clot [42,144,145,147,148]. According to a study, conducted in APP/PS1 transgenic mice, an AD model, an increase in fibrin within blood vessels and the abnormal accumulation of the extracellular matrix in the brain tissue were found to induce neurovascular damage and neuroinflammatory responses [148]. The progression of Aβ deposition in the brain tissue was accompanied by vascular damage and inflammatory responses [148], which, in turn, accelerated the progression of AD due to the excessive accumulation of fibrin [148]. Thus, reducing the abnormal accumulation of fibrin in the extracellular matrix and brain vasculature may mitigate brain damage and slow down disease progression [148]. Ryu et al. showed that treatment with the 5B8 antibody targeting the fibrin epitope γ377-395 prevented abnormal fibrin accumulation in a 5XFAD AD animal model, resulting in decreased neuroinflammation and neuronal damage [42].
2.4. Other Proteins and Factors
In addition to coagulation factors, blood vessels contain a diverse array of proteins and factors that serve a range of functions beyond their role in hemostasis [83,84,85,86,149,150,151,152,153,154,155]. These proteins and factors can also infiltrate the brain and have harmful effects [149,150,151,152,153,154,155]. In one study, the blood-borne Aβ protein was found to penetrate the BBB and accumulate in the microglia, leading to AD pathogenesis [155]. The blood Aβ protein induces neuroinflammation and microglial activation, leading to the formation of brain deposits and neuronal damage [155]. AD patients have increased levels of albumin and immunoglobulin G (IgG) in their blood compared to normal individuals, and the passage of these proteins through the impaired BBB into the brain may play a role in the pathophysiology of AD [149,151,154]. Additionally, the brain tissues of patients with AD patients were shown to have increased concentration of hemoglobin-derived peptides, which may contribute to neuroinflammation and neuronal damage [153]. Another study reported increased iron levels in the hippocampal region of patients with AD using magnetic resonance imaging, which may be related to the pathophysiological changes in AD [152]. Plasminogen derived from blood was shown to induce Aβ accumulation in the brain through microglial activation, further aggravating the neuroinflammatory responses [150]. Specifically, plasminogens derived from blood could induce Aβ accumulation in the brain through microglial activation, leading to further promotion of neuroinflammatory responses [150]. Therefore, further investigation is required to examine additional proteins and factors present in the blood and to investigate their mechanisms of infiltration into the brain and activation of microglial cells.
In addition, the overexpression or overactivation of certain brain proteins could disrupt BBB integrity. A study investigated the role of IL-1β in regulating BBB permeability and found that IL-1β activated microglia, leading to damage and inflammation of vascular endothelial cells; this, subsequently, affected angiogenesis and increased vascular permeability [156]. In addition, an investigation of BBB destruction and cell death in an ischemic stroke model revealed that the TNF-α derived from microglia induced neuronal death, exacerbating BBB destruction [157]. Some studies revealed that blood-derived extracellular vesicles, such as microvesicles, transported into the brain could influence microglial activation, and extracellular vesicles generated through microglial activation could induce neuronal damage in the AD brain [158,159]. Moreover, High Mobility Group Box-1 (HMGB1) was identified as one of the key factors that promotes BBB breakdown. HMGB1 increases in the setting of brain injury or inflammation, inducing an inflammatory response in the endothelial cells of the BBB and increasing BBB permeability [160]. Matrix metalloproteinases (MMPs) are protein-degrading enzymes that regulate the extracellular matrix, but excessive MMP activity can cause brain tissue damage and inflammation [160,161]. Increased activity of MMP-9 was shown to be involved in the breakdown of the BBB [161]. Therefore, further research on the signaling pathways such as MMP-9 and other proteins in relation to BBB damage may support the development of new therapeutic strategies for AD.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines11051383
This entry is offline, you can click here to edit this entry!