Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Junichi Fujii | -- | 4461 | 2023-05-16 10:36:55 | | | |
| 2 | Jessie Wu | + 9 word(s) | 4470 | 2023-05-16 10:58:37 | | | | |
| 3 | Jessie Wu | + 1 word(s) | 4471 | 2023-05-16 11:00:09 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Fujii, J.; Osaki, T.; Soma, Y.; Matsuda, Y. GSH Protects Cells against Stress through Multiple Pathways. Encyclopedia. Available online: https://encyclopedia.pub/entry/44353 (accessed on 09 August 2026).
Fujii J, Osaki T, Soma Y, Matsuda Y. GSH Protects Cells against Stress through Multiple Pathways. Encyclopedia. Available at: https://encyclopedia.pub/entry/44353. Accessed August 09, 2026.
Fujii, Junichi, Tsukasa Osaki, Yuya Soma, Yumi Matsuda. "GSH Protects Cells against Stress through Multiple Pathways" Encyclopedia, https://encyclopedia.pub/entry/44353 (accessed August 09, 2026).
Fujii, J., Osaki, T., Soma, Y., & Matsuda, Y. (2023, May 16). GSH Protects Cells against Stress through Multiple Pathways. In Encyclopedia. https://encyclopedia.pub/entry/44353
Fujii, Junichi, et al. "GSH Protects Cells against Stress through Multiple Pathways." Encyclopedia. Web. 16 May, 2023.
Copy Citation
Methionine (Met) and cysteine (Cys) are the only sulfur-containing amino acids, and Cys can be synthesized through the transsulfuration reaction associated with Met metabolism. Met is metabolized to S-adenosylmethionine (SAM), which provides either the carbon backbone for polyamines or methyl groups for some other compounds, including DNA . Meanwhile, Cys comes from extracellular sources or is the product of the transsulfuration reaction and then becomes the precursor for sulfur-containing components or mediators. These include glutathione (GSH), taurine, coenzyme A, hydrogen sulfide, iron–sulfur [Fe–S] cluster, and persulfides.
xCT
glutathione peroxidase
glutaredoxin
γ-glutamyl transferase
1. Reductive Detoxification of Peroxides via Glutathione Peroxidases
The oxidation of glutathione (GSH) occurs by the direct reaction with oxidants, but this is not a very efficient process. The reduction of peroxides to the corresponding alcohols is effectively catalyzed by glutathione peroxidases (GPX), which results in GSSG [1]. Whereas certain other proteins, such as catalase and peroxiredoxin, exhibit peroxidase activities, GPX is a dominant peroxidase family that is made up of eight members in mammals. Because GPX is constitutively active within cells and requires GSH other than substrate for activity, the supply of GSH determines the GPX activity. GPX1 to GPX4 possess a selenocysteine (Sec) instead of a Cys in their catalytic center (Figure 1). Replacing Sec with Cys decreases the catalytic efficiency of this molecule by less than 1%. Whereas Sec-containing wild-type GPX4 is capable of conferring resistance to irreversible overoxidation by peroxides [2], excessive hydrogen peroxide inactivates GPX1 by oxidatively converting Sec to dehydroalanine in human erythrocytes [3]. A diet with selenium deficiency results in a decrease in their activity due to the impaired synthesis of Sec. Whereas GPX largely functions to reduce hydrogen peroxides by means of the donation of an electron from GSH, GPX4 reduces phospholipid hydroperoxides to their alcohol forms and, hence, can suppress ferroptosis [4][5]. Since ferroptosis is considered to occur under a variety of physiological and pathological conditions, the suppression of lipid peroxidation by GPX4 has attracted much attention [6][7]. The genetic ablation of GPX4 causes embryonic lethality in mice, which can be overcome by vitamin E (α-tocopherol) supplementation [8], whereas this is not the case in mice lacking other GPX genes. Peroxiredoxin (PRDX) is another large family of proteins with peroxidase activity. PRDX family members mostly exhibit thioredoxin-dependent peroxidase activity, whereas PRDX6 exceptionally exhibits GSH-dependent peroxidase activity towards lipid peroxides. Thus, the enzymatic properties of PRDX6 are similar to those of GPX4, but there is essentially no similarity in their structure [9]. Since numerous studies have reported on the roles of GPX in neuronal systems, readers are referred to other review articles for details concerning the reactions and physiological significance of the GPX family [1][7][10]. The decline in the GPX activity increases peroxides, which, in the presence of iron, results in the production of hydroxyl radicals that cause oxidative DNA damage. Then, cells with DNA damage may undergo tumor development [4]. Oxidative stress-induced tumorigenesis is a major research subject in itself and is discussed repeatedly, so researchers will not discuss it further in this research.
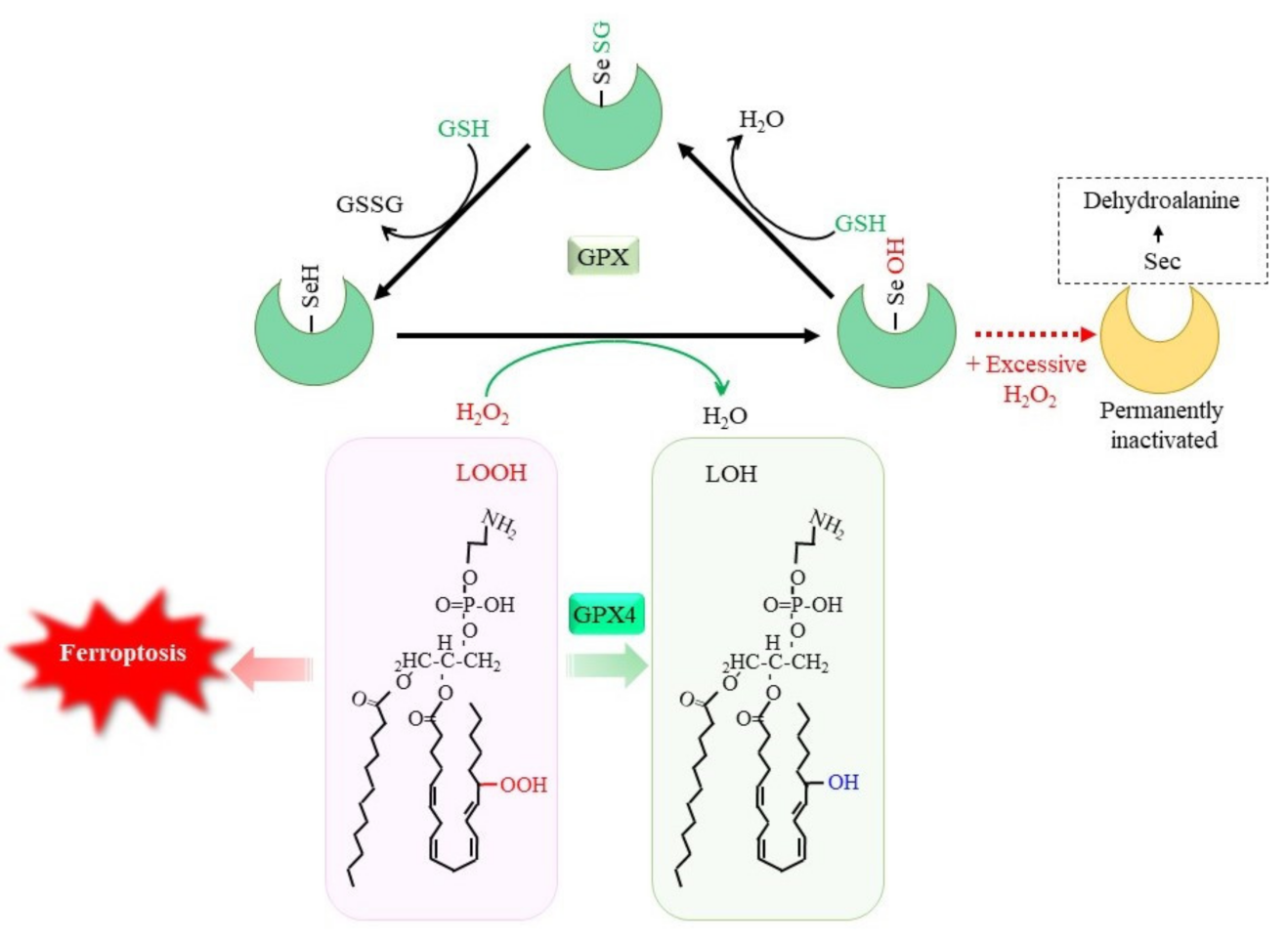
Figure 1. GSH-dependent reduction of peroxides by glutathione peroxidase (GPX). Selenocysteine (Sec) constitutes the catalytic center of GPX1 to GPX4, among GPX family members. The reaction of GPX with peroxides converts SeH to SeOH in the Sec residue, which transiently leads to glutathionylation. Reaction of another GSH regenerates SeH and releases GSSG. Excessive hydrogen peroxide converts Sec to dehydroalanine, leading to permanent inactivation of GPX1. Accumulation of phospholipid hydroperoxides causes ferroptosis, whereas GPX4 specifically reduces them to the alcohol form, leading to cell survival. LOOH, lipid hydroperoxide; LOH, lipid alcohol.
2. Rescuing Proteins from Oxidative Modification through Glutaredoxin
Whereas SH groups can exist in four oxidized states: disulfide, sulfenic acid, sulfinic acid, and sulfonic acid, under an oxidative environment, the disulfide and sulfenic acid forms can generally be reduced by a biological redox system [11]. Cys–SH groups of proteins can form mixed disulfides with their own SH group or with SH groups from other molecules under oxidizing conditions (Figure 2A). Since GSH is the most abundant thiol, a mixed disulfide with GSH, called S-glutathionylation, occurs preferentially. This modification either inhibits or activates the enzyme, depending on the position of the Cys residue within the protein [12]. Approximately 1% of total glutathione is present as the mixed disulfide form with proteins in the normal liver, and the amount of protein-bound glutathione reaches 20–50% upon an oxidative insult [13]. The glutathione thiyl radial and GSNO form the S-glutathionylation of proteins in an accelerated manner. S-glutathionylation proceeds preferentially under oxidative conditions, but is not always harmful, because the formation of a mixed disulfide prevents further oxidation of the SH group [14]. GRX has been identified as a GSH-dependent reductase of disulfides in ribonucleotide reductase during its catalytic cycle [15][16] and, hence, acts as an alternate electron donor for TRX [12]. Because S-glutathionylation protects Cys–SH groups in proteins from further oxidation and since they can be reduced back, this post-translational modification is regarded as a type of protective mechanism for essential SH groups in proteins under oxidative stress [17]. GRX can reduce proteins with intrinsic disulfide bonds or S-glutathionylated proteins back to the native conformation by employing GSH, which releases GSSG. Mammals produce two GRXs, GRX1 and GRX2 [16][17]. Whereas GRX1 resides mainly in the cytoplasm, it can be translocated into the nucleus upon certain stimuli. GRX2 is present in two forms, namely, a mitochondrial and a nuclear GRX.
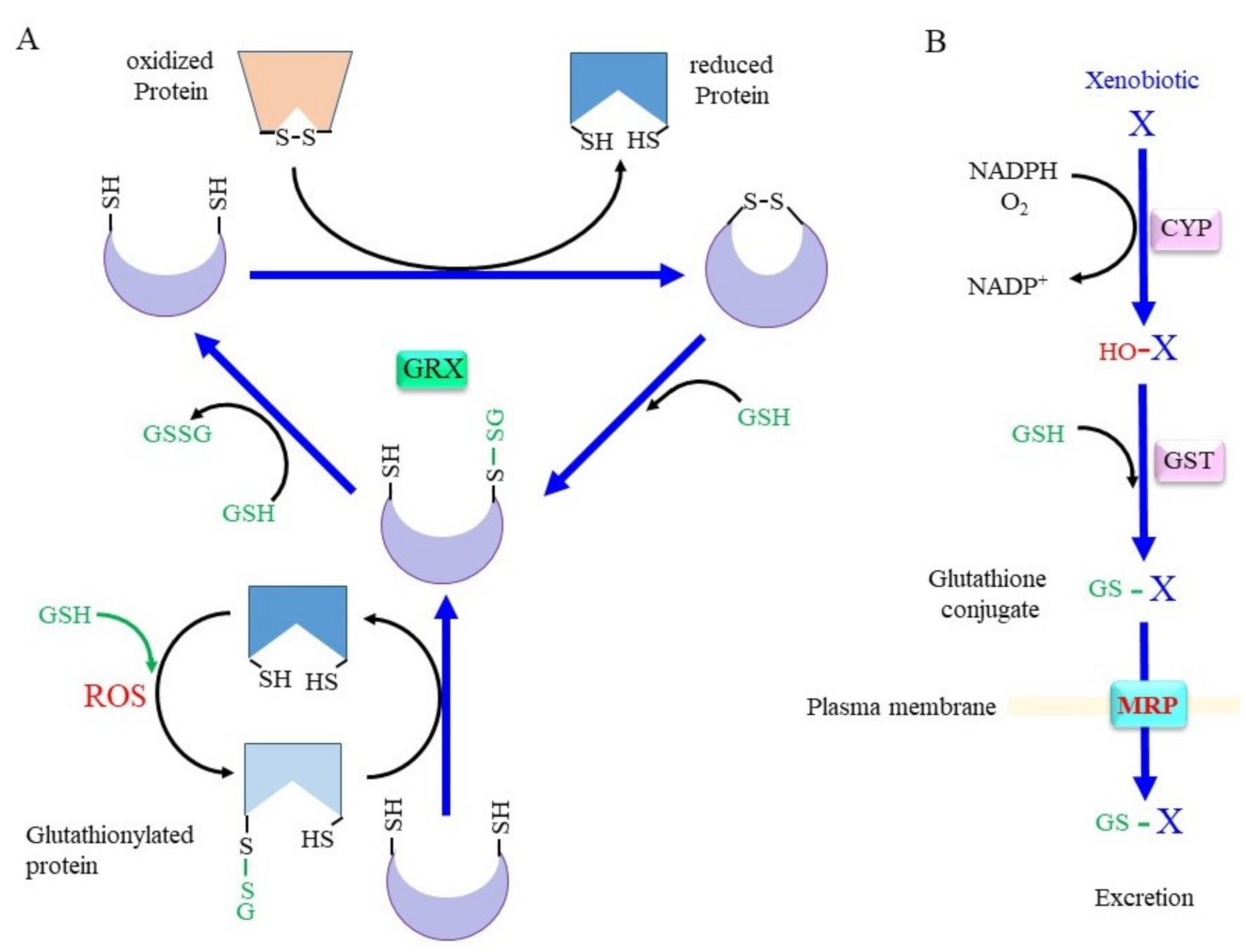
Figure 2. Scheme for the conjugation of glutaredoxin (GRX) and GSH. (A) The upper scheme of the reaction shows the reduction of oxidized proteins, and the lower scheme shows GRX accepting the GSH moiety from glutathionylated proteins. When the protein with a disulfide bond reacts with reduced GRX, the protein is reduced instead and GRX is oxidized. Upon reaction with GSH, one cysteine (Cys) residue of the oxidized GRX becomes an SH group and that of the other Cys becomes glutathionylated. Reaction of glutathionylated GRX with another GSH results in fully reduced GRX and GSSG. (B) Xenobiotics and hydrophobic metabolite (X) first undergo hydroxylation by oxygenases such as cytochrome P450 oxidases (CYP). Then, glutathione S-transferase (GST) catalyzes GSH conjugation. Resulted glutathione conjugates are exported via multidrug resistance regulator (MRP). ROS, reactive oxygen species.
Redox regulation by the GSH/GRX system plays a pivotal function in the central nervous system [18][19]. The excitatory amino acid L-β-N-oxalylamino-L-alanine (L-BOAA) causes corticospinal neurodegeneration in humans and the loss of GSH in mice. The mitochondrial electron transfer complex (ETC)-I in the motor cortex is selectively lost by L-BOAA, but GRX appears to protect ETC-I from degradation [20]. Estrogen may be involved in preserving higher levels of GRX in certain regions of the central nervous system and, in female mice, protects them against mitochondrial dysfunction caused by L-BOAA [21]. GRX1 helps to maintain mitochondrial integrity and prevents the loss in mitochondrial membrane potential caused by L-BOAA [22]. 6-Hydroxydopamine is easily oxidized and results in the formation of the quinone form, which is a highly reactive species and a powerful neurotoxin [23]. Both the TRX and the GRX systems directly mediate the reductive detoxification of 6-hydroxydopamine quinone and protect neurons from dopamine-induced cell death.
GRX1 reportedly regulates the protein levels of DJ-1 in the midbrain of mice [24]. The mitochondrial form of GRX2 as well as TRX1 contributes to neuronal integrity during hypoxia [25] and protects neuronal cells against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-mediated mitochondrial dysfunction [26]. The downregulation of GRX1 leads to dopaminergic degeneration and PD-relevant motor deficits in mice [27]. Exogenously added cell-permeable PEP-1-GLRX1 also suppresses the dopaminergic neuronal cell death induced by MPTP [28]. GRX2 may also be involved in the function of mammalian dopaminergic cells and oligodendrocytes through the biogenesis of [Fe–S] clusters [29][30]. Increased levels of GRX1 are associated with the early onset of PD in patients, which suggests that upregulated GRX1 promotes neuroinflammation and leads to the development of PD [31]. However, other studies have reported decreased levels of GRX in PD patients [32]. Given the roles of GRX in protein thiol homeostasis, the upregulation of GRX1 is rather considered to be result of a compensatory reaction against oxidative protein modification.
The oxidation of F-actin caused by amyloid-β (Aβ) is diminished by GRX1 in Alzheimer’s disease (AD) model mice [33]. A decrease in GRX1 levels may lead to synaptic dysfunction during AD pathogenesis by directly disrupting the F-actin architecture in spines. On the contrary, Aβ may also exert neurotoxicity in AD through oxidizing GRX1 or TRX1 [34]. In familial ALS patients, the aggregation of mutant SOD1 is a proposed cause for the degeneration of motoneurons. Whereas the overexpression of GRX1 increases the solubility of mutant SOD1 in the cytosol, this does not alleviate mitochondrial damage in SH-SY5Y cells. However, the overexpression of GRX2 increases the solubility of mutant SOD1 in mitochondria and preserves mitochondrial function, which results in neuronal cells being protected from apoptosis [35]. Thus, GRX, with the help of GSH, can prevent the development of major neurodegenerative diseases such as PD, AD, and ALS.
3. Glutathione Conjugation in the Detoxification of Xenobiotics and in the Production of Bioreactive Compounds
Conjugation with GSH, glucuronate, and sulfate are catalyzed by GST, UDP-glucuronate transferase, and sulfotransferase, respectively, and constitute three major detoxification systems for xenobiotics and intrinsic compounds that are highly hydrophobic [36][37]. Oxygen molecules are usually introduced into hydrophobic compounds by the action of cytochrome P450 oxidases (CYP) before glutathione conjugation by GST (Figure 2B). In addition to GSH conjugation, GST also exhibits GSH-dependent peroxidase activity, albeit with much less efficiency compared to GPX. The mouse has 21 GST genes, and many of these genes are genetically knocked out, as reported in previous studies [38][39][40]. Many polymorphisms are found in GST genes that are likely risk factors for PD [41]. Whereas glucuronidation and sulfate conjugation are generally thought to have largely beneficial effects, excessive GSH conjugation may cause redox imbalance and damage cells due to the consumption of GSH, as is typically observed in the case of livers with an acetaminophen overdose [42].
On the other hand, GSH is used as a building block or cysteine donor for the synthesis of cysteinyl leukotrienes (CysLT) that include LTC4, LTD4, and LTE4 (Figure 3A). CysLT is the active component of a slow-reacting substance that causes anaphylaxis, the contraction of smooth muscle, and an increase in vascular permeability [43]. CysLT is also an inflammatory lipid mediator that is involved in the pathophysiology of respiratory diseases and may also be associated with defects in the central nervous system, including cerebral ischemia, epilepsy, and AD [44]. Regarding the synthetic pathway, 5-lipoxygenase first catalyzes the formation of an arachidonate epoxide, and GST family members (MGST2, 3, and GSTM4), as well as LTC4 synthetase, then catalyze the conjugation of GSH to the epoxide [45]. After their secretion from cells, GGT1 or GGT5 hydrolytically removes the γ-glutamyl group of LTC4, which results in the formation of LTD4. Extracellular peptidases finally catalyze the hydrolytic removal of the Gly unit from LTD4, and this results in the production of LTE4. CysLT acts through G protein-coupled receptor subtypes that are referred to as CysLTR-1 and CysLT-2 and are present on neurons, astrocytes, microglia, and vascular endothelial cells in the brain [46]. Moreover, proteins, such as G protein-coupled receptor 17 (GPR17), G protein-coupled receptor 99 (GPR99), and peroxisome proliferator-activated receptor-γ (PPARγ), may also act as receptors for CysLT. Since CysLT is also involved in inflammatory responses, its excessive production may result in neuronal tissue damage.
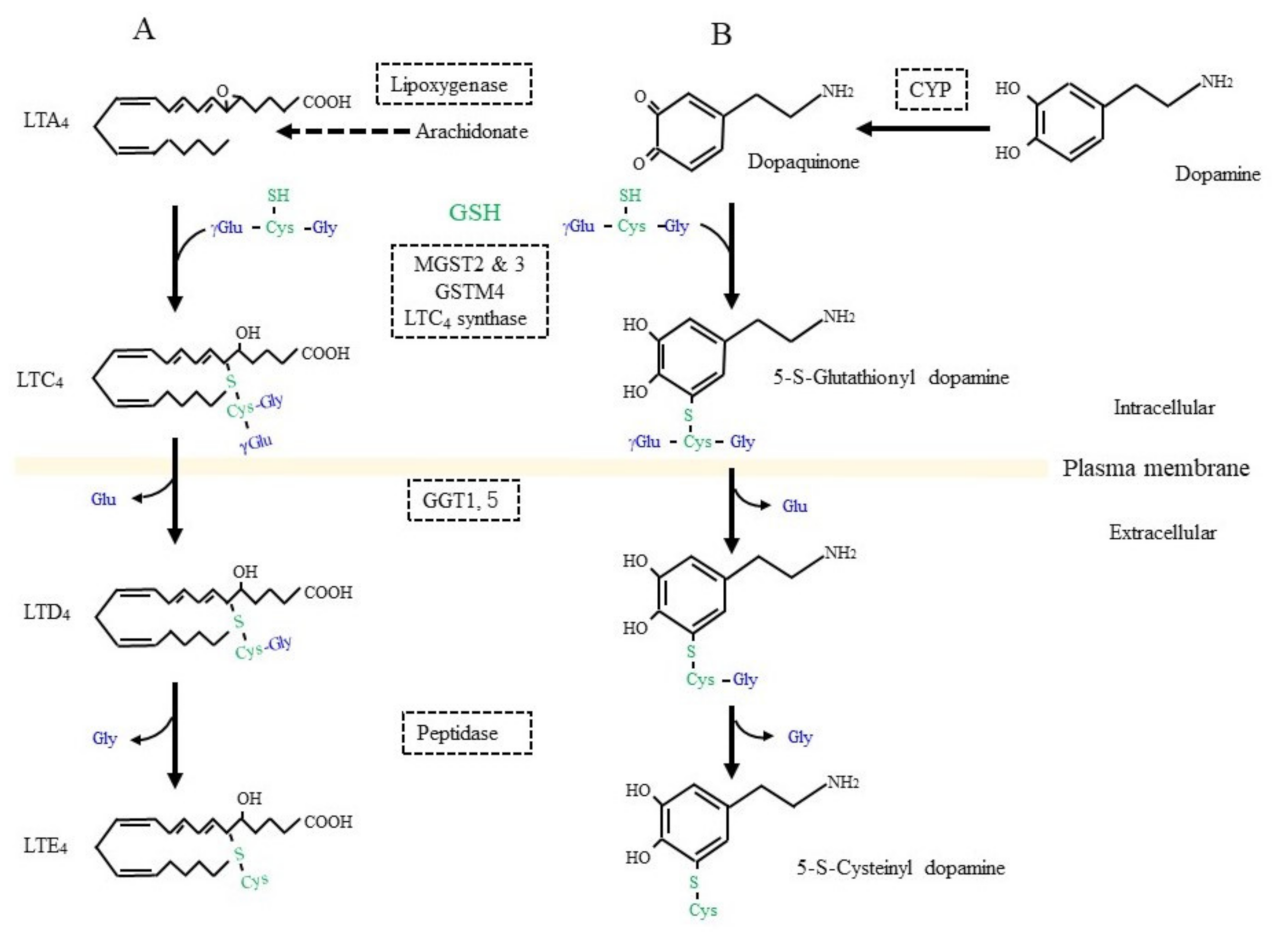
Figure 3. GSH as a building block for some bioreactive compounds. (A) GSH is conjugated to leukotriene (LT) A4 by the catalytic action of LTC4 synthetase or some other glutathione S-transferase (GST) isozyme. γ-Glutamyl transferase (GGT) hydrolytically removes the γ-glutamyl moiety from LTC4, which enables access to a peptidase that removes the glycine (Gly) unit from LTC4 and results in LTD4 formation. (B) Dopamine is oxidized to dopaquinone by oxidases such as cytochrome P450 (CYP). After conjugation with GSH, 5-S-cysteinyl dopamine is produced by the action of similar enzymatic process to those for LTD4 synthesis.
4. Glutathione S-Conjugates, and S-Nitrosoglutathione (GSNO) in Nitric Oxide Signal Transduction
Nitric oxide (NO) that is produced through both enzymatic reactions and non-enzymatic reactions exerts a variety of beneficial functions, including the relaxation of the vasculature and the modulation of neurotransmission, whereas the presence of abundant levels may impair the redox balance [47]. Whereas the iron ion, notably ferrous iron, is the preferred target of NO, SH groups in amino acids and proteins are also reactive and are targets, which results in the formation of S-nitrosothiol (SNO). Since reactive SH tends to play a primary role in a redox reaction that includes TRX and the ubiquitin system [48], and non-protein thiols, including coenzyme A [49], S-nitrosylation is a pivotal post-translational modification that is involved in cellular signaling. Excessive nitrosylation of target proteins may cause dysfunction, aberrant activation of physiological processes, and ultimately cell death. S-Nitrosylation occurs in many different PD-related proteins, including peroxiredoxin 2, XIAP, and PDI [50]. The balance between S-nitrosylation and denitrosylation determines whether SNO acts as a signaling mechanism or causes nitrosative stress [51].
GSNO is dominantly produced due to the abundant presence of GSH in cells and may act as the donor for trans-nitrosylation reactions [52]. GSNO is transported out of cells via MRP [53]. GSNO-reducing activities, which are intrinsic to formaldehyde dehydrogenase (GSNOR), class III alcohol dehydrogenase (ADH5), and TRX/thioredoxin reductase, play pivotal roles in moderating GSNO levels [54]. ADH5 appears to be a major GSNO reductase that acts in an NADH-dependent manner [55]. Whereas carbonyl reductase 1 (CBR1) preferentially reduces GSNO [56][57], a form of aldehyde reductase AKR1A1 also exerts GSNO reductase activity with the formation of glutathione-sulfinamide derivatives [52]. A GSNOR deficiency induces the S-nitrosylation of focal adhesion kinase 1 (FAK1), which results in the enhanced autophosphorylation of FAK1 and tumorigenicity being sustained [58]. Since detailed information concerning the action of S-nitrosylation is not the focus of this research, readers are directed to recent review articles that are associated with neurodegenerative diseases [59][60].
5. Glutathione Status Associated with Neuronal Diseases
Researchers briefly revisit the relationship between major neurological diseases and GSH status in this research. A GSH deficiency is associated with various neurological disorders, including neurodegenerative diseases, ischemic disease, schizophrenia, and tumors [61][62][63][64]. Ferroptosis, which is associated with declined GSH, is assumed to be involved in neurodegenerative diseases, including AD, PD, and ischemic disease [62][64][65]. Because polyunsaturated fatty acids (PUFA) are rich in the brain and are susceptible to being peroxidized, GPX4 is predominantly present and protects neurons from ferroptosis. Here researchers outline three typical types of neurodegenerative diseases, AD, PD, and ALS, in association with GSH metabolism.
AD is the most common neurodegenerative disease. Aggregation of the Aβ peptide eventually causes AD by inducing neuronal cell death. The hallmarks of AD include elevated ROS levels and the enhanced production of lipid peroxidation products, decreases in GSH and GPX4, and the accumulation of iron, which are also hallmarks of ferroptosis [66][67]. The administration of desferrioxamine, an iron chelator, to AD patients reportedly leads to a significant reduction in the rate of decline of daily living skills, suggesting that sustained iron chelation may be beneficial in slowing the progression of this disease [68]. The administration of α-tocopherol, which suppresses ferroptosis by inhibiting lipid peroxidation, was also reported to moderate AD in patients [69]. Moreover, treatment with NAC was reported to prevent cognitive impairment in an AD model mouse that was induced by intracerebroventricularly administered streptozotocin [70]. The injection of Aβ oligomers into the CA3 hippocampal region of the rat brain triggers synaptotoxic effects that are represented by abnormal Ca2+ signals and mitochondrial dysfunction, whereas feeding NAC for 3-weeks prior to Aβ injections prevented these deleterious effects [71]. It is therefore considered likely that ferroptosis is related to the onset or exacerbation of AD, and that the Cys–GSH axis exerts protective action in preventing neuronal cell death. Using Drosophila models, a close correlation between changes in GSH redox potential with AD disease onset caused by Aβ and progression was observed [72].
Dopamine is a pivotal neurotransmitter, and its deficiency is a cause for PD, which is also a common neurodegenerative disease, next to AD. Progressive dopaminergic neuronal loss in the substantia nigra pars compacta is the characteristic pathology of PD patients. Since iron accumulation is associated with PD, ferroptosis as well as oxidative stress are also considered to be possible causes [73][74][75]. Dopamine can be converted into dopamine-o-quinone in the oxidative pathway, which is efficiently catalyzed by the presence of metal ions such as iron, copper, and manganese, as well as by the action of ROS-producing enzymes, such as xanthine oxidase, cytochrome P450, prostaglandin H synthase, and lactoperoxidase [76]. The metabolism of GSH in association with dopamine is deeply involved in this fatal disease [77][78]. Several enzymes, including GST family members and LTC4 synthase, catalyze the nucleophilic addition of GSH to dopamine-o-quinone, which results in the formation of 5-S-glutathionyl dopamine (Figure 3B). MRP then appears to export 5-S-glutathionyl dopamine [79]. GGT that is present on the extracellular surface of astrocytes then catabolizes the removal of the γ-glutamyl group from 5-S-glutathionyl dopamine [80]. The Gly unit is finally removed by the action of an extracellular peptidase. The resulting Cys-dopamine is toxic to neurons in the substantia nigra pars compacta and may lead to neuronal death. In addition to the neuronal toxicity of these compounds, Cys/GSH consumption may impair GPX4 function and predispose neurons to ferroptosis. Similar processing occurs on glutathione-conjugated acetaminophen, and the resulting cysteinyl-acetaminophen is reportedly a major contributor to renal toxicity [81].
Despite the fact that it is much less widespread compared to AD and PD, ALS affects motor neurons in the cerebral cortex, brainstem and spinal cord and leads the death rate due to respiratory failure within five years. Oxidative cell damage also appears to be involved in the development of ALS, and, hence, the GSH redox system is considered to exert a beneficial action [82]. In fact, the modification of the Cys residue in either GSH or in proteins occurs and may be involved in the pathogenesis of ALS [83]. Whereas the significance of the Cys–GSH axis has been implied, clinical trials that include the administration of GSH, Cys or procysteine have not been successful. Meanwhile, the administration of edaravone, a radical scavenger, to rats was reported to alleviate spinal cord injury [84]. Edaravone is now a licensed radical-scavenging drug for the treatment of ALS as well as strokes [85][86]. These collective results suggest that radical scavenging rather than fueling the Cys–GSH axis might be more advantageous in slowing the progression of ALS.
6. Production of a Variety of γ-Glutamyl Peptides by Means of γ-Glutamyl–Cysteine Synthetase and γ-Glutamyl Transferase
A variety of peptides and amino acids, which may not always be composed of proteinous amino acids, are present in blood plasma and tissues including the brain (Figure 4). Taurine is synthesized from Cys with cysteine dioxygenase as the rate-limiting enzyme [87]. Astrocytes predominantly produce taurine, which exerts pleiotropic actions in the central nervous system [88]. The levels of taurine and 2-hydroxybutyrate, a metabolite of the transsulfuration pathway, are elevated in plasma and cells from sporadic ALS patients, suggesting that the pathogenesis of ALS is associated with metabolic stress [89]. Because taurine does not contain a carboxyl group, it presents largely in the free form and constitutes the second amino acid in dipeptides. γ-Glutamyl taurine (γ-Glu-Tau) is produced in the brain, and is reportedly mediated by GGT [90]. In fact, a variety of reactive peptides that include γ-glutamyl peptides have been reported to be present in the brain. Researchers recently established a liquid chromatography–mass spectrometry (LC–MS)-based assay method that can provide structural information on the products of the enzymatic reaction of γ-GCS and GGT. The results indicate that, despite the high Km of taurine for the γ-GCS reaction, its abundant presence indeed also enables the generation of γ-Glu-Tau by γ-GCS [91][92]. The resulting γ-Glu-Tau may interact with excitatory aminoacidergic neurotransmission [93] and exert antiepileptic actions [94], although the nature of its function remains ambiguous [95]. GGT and γ-GCS appear to be the enzymes that are responsible for the production of various γ-glutamyl peptides, although the amount and diversity of γ-glutamyl peptides are small in the normal mouse brain compared to the liver and kidney [92]. These observations imply that the concentrations of Cys are properly maintained in the brain under healthy conditions. Aberrant syntheses of γ-glutamyl peptides other than GSH by γ-GCS and/or GGT reactions can also be a predictive marker for the condition of the central nervous system in cases of a Cys/GSH deficiency.
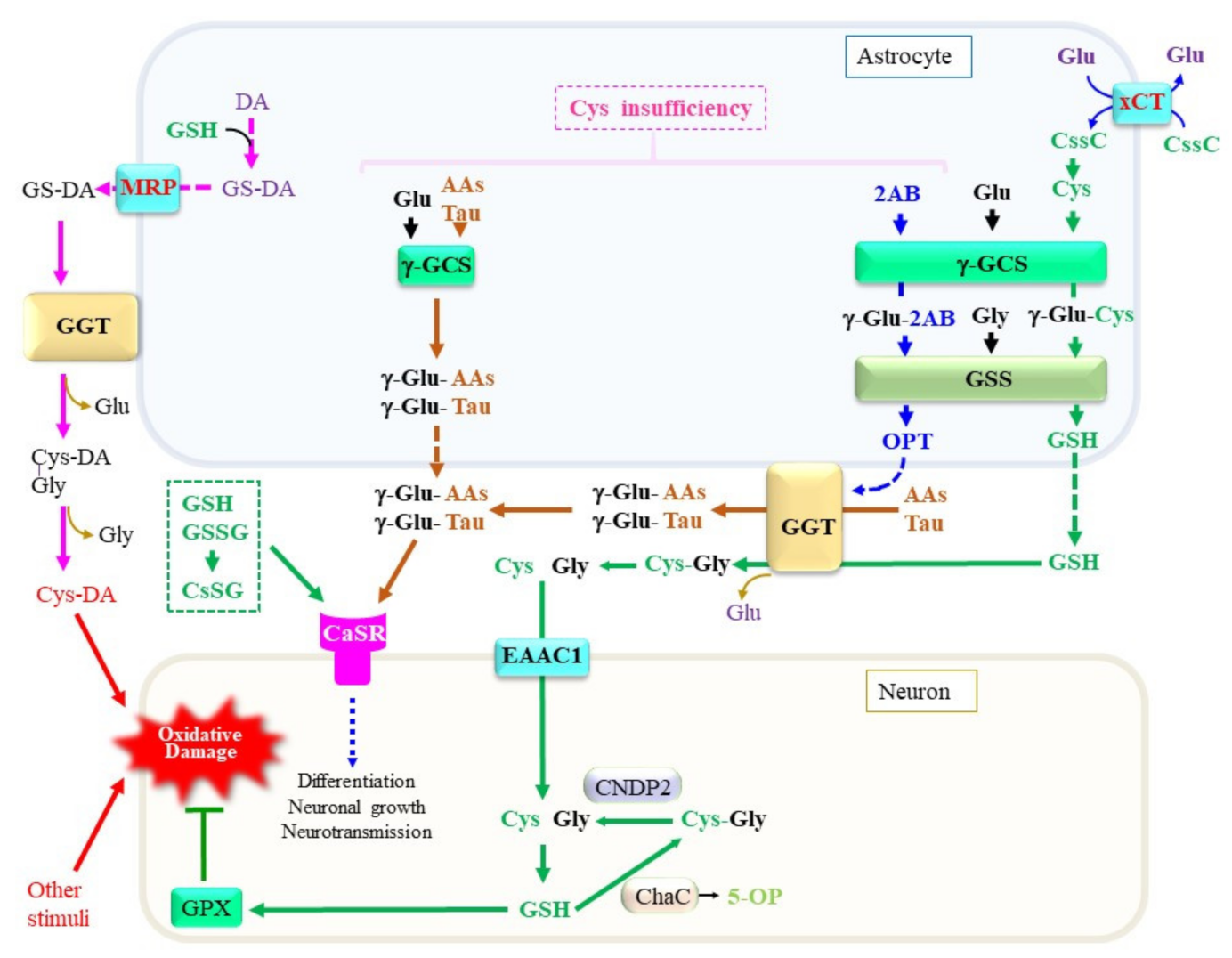
Figure 4. Production, metabolism, and cellular signaling of γ-glutamyl peptides. Due to the diversity of the nervous system, the metabolic pathways shown in this diagram are only examples of a certain central nervous system and may not be applicable to all neuronal cells. When Cys is present at sufficient levels, GSH is produced. However, under a Cys insufficiency, high levels of 2-aminobutryrate (2AB) are synthesized through the transsulfuration pathway, followed by a transaminase reaction, and it becomes a substrate for γ-GCS, which may lead to the production of ophthalmate (OPT). When other amino acids, including taurine, are used instead of Cys, a variety of γ-glutamyl peptides such as γ-Glu-Tau are produced by γ-GCS reactions inside cells. γ-Glutamyl transferase (GGT) on astrocytes either hydrolytically removes the γ-glutamyl moiety of extracellular GSH and γ-glutamyl peptides or transfers their γ-glutamyl moiety to other amino acids (AAs) to generate new γ-glutamyl peptides. The resultant Cys–Gly dipeptide can be hydrolyzed to its constituent amino acids by the action of a dipeptidase. The function of the calcium-sensing receptor (CaSR) may be modulated by the binding of these γ-glutamyl peptides. AAs, any amino acid; DA, dopamine. γ-GCS, γ-glutamyl-cysteine synthetase; GSS, glutathione synthetase; ChaC, γ-glutamylcyclotransferase; MRP, multidrug resistance regulator; CNDP2, carnosine dipeptidase 2; EAAC1, excitatory amino acid carrier 1; GPX, glutathione peroxidase; DA, dopamine; Cys-DA, cysteinyl-dopamine; xCT, cystine transporter.
Attempts to elucidate the roles of γ-glutamyl peptides in the brain have just begun, and available information concerning them is limited. Researchers therefore discuss this issue using the liver as an example because it is the most extensively investigated organ. During the production of Cys through the transsulfuration pathway, 2-oxobutyrate is produced as a result of the CSE-catalyzed cleavage of cystathionine [96]. The resulting 2-oxobutyrate is converted into 2-aminobutyrate (2AB) by transferring an amino group from Glu via aminotransferases. Because 2AB is also a preferred substrate for γ-GCS next to Cys, γ-Glu-2AB is produced under conditions of a Cys insufficiency. Whereas γ-GCS is suppressed by physiological levels of GSH via a feedback mechanism, the consumption of GSH stimulates γ-GCS activity [97]. When γ-GCS utilizes 2AB as the acceptor substrate, γ-Glu-2AB is produced and is then converted to γ-Glu-2AB-Gly by the GSS reaction, which is denoted as ophthalmic acid (OPT) [98]. The production of excess levels of OPT is observed in the mouse liver under conditions of a Cys deficiency, typically upon an acetaminophen overdose [99]. Starvation of mice causes an insufficient supply of amino acids, including Cys and Met, which also leads to an increase in OPT production in the blood plasma [100]. Intriguingly, several types of γ-glutamyl peptides were reported to be elevated in the blood plasma of patients who are suffering from liver diseases [101]. Since the liver is a central organ for amino acid metabolism, hepatocytes may suffer oxidative stress and result in the consumption of both GSH and Cys. It is conceivable that, under these conditions, activation of the transsulfuration pathway produces 2-oxobutyrate along with Cys. However, Cys is rapidly recruited for GSH synthesis, which consequently stimulates the utilization of 2AB for the production of OPT. NAC is generally used in the treatment of an acute liver injury caused by an acetaminophen overdose. For similar reasons, NAC or its lipophilic derivatives increase the levels of cellular Cys and, consequently, GSH, which then may exert therapeutic effects on neuronal diseases [102].
Ferroptosis is reportedly suppressed by producing not only GSH but also other γ-glutamyl peptides by γ-GCS [103]. The anti-ferroptotic effects of the production of γ-glutamyl peptides other than GSH cannot be explained by the reductive detoxification of lipid peroxides via GPX4. Because the inhibition of γ-glutamyl peptide synthesis elevates cellular Glu levels, the stimulation of the Glu metabolism appears to be the likely mechanism for executing ferroptosis under a Cys insufficiency. This notion is consistent with findings that elevations in electrochemical potential in mitochondria are associated with ferroptosis induced by Cys deprivation [104]. Because actively proliferating cells are more sensitive to ferroptotic stimuli, the activation of Glu-centered carbon metabolism likely produces more radical species, which may stimulate lipid peroxidation reactions and consequent ferroptosis [105]. Meanwhile, the cysteine-sparing effect of taurine has been proposed in hepatocytes [106], although taurine cannot directly compensate for a Cys deficiency. These collective data can also be interpreted to indicate that the formation of γ-Glu-Tau and other γ-glutamyl peptides by either intracellular γ-GCS or extracellular GGT prevents the excitatory cytotoxicity caused by excessive levels of Glu.
7. Calcium-Sensing Receptor as a Target of γ-Glutamyl Peptides
In addition to GSH, many γ-glutamyl peptides have been detected, and the γ-glutamyl moiety of N-terminal amino acids stabilizes them. The physiological significance of this peptide-specific modification has long been debated. The pharmacological benefit of supplementation of γ-Glu-Cys has recently been proposed under pathological conditions, such as strokes [107], inflammation [108][109][110][111], ALS [112], and ischemia/reperfusion injury [113]. The pharmacological action of γ-Glu-Cys appears to be largely attributable to an increased production of GSH [114][115][116]. Although the direct donation of an electron from γ-Glu-Cys to GPX1 has been also demonstrated [117], the presence of GSS mostly converts γ-Glu-Cys to GSH. Therefore, the use of intrinsic γ-Glu-Cys as the substrate for GPX1 may occur in limited organs such as the kidney, which uniquely contains four times more γ-Glu-Cys than GSH [92].
Extracellular levels pf GSH, GSSG, and mixed disulfides between Cys and GSH (CySSG) can modulate the function of the G-protein-coupled calcium-sensing receptor (CaSR) [118][119]. CaSR is systemically expressed, including in the brain and intestine, and a primary function of CaSR appears to be maintaining extracellular calcium ion levels within a physiological range, 1.1–1.3 mM, by regulating the secretion of the parathyroid hormone [120]. CaSR is abundantly expressed in the circumventricular regions and substantially within the hippocampus, hypothalamus, and striatum. Concerning the roles of CaSR in the neuronal regulation of nutrition, several γ-glutamyl peptides appear to bind CaSR. The extracellular domain of this protein contains a binding pocket for GSH-related compounds [119]. The allosteric activation of CaSR by γ-Glu-Cys was reported to suppress inflammation in colitis model mice [108]. CaSR expressed within the gastrointestinal tract plays roles as a mediator of “kokumi taste” modulation and is responsible for regulating the release of dietary hormones in response to amino acids in the intestine [121][122].
The roles of CaSR in neurons reportedly include neuronal growth, migration, differentiation, and neurotransmission [120]. It has been proposed that CaSR has a critical role in the central neuronal system under pathological conditions, such as ischemia, AD, and in neuroblastomas [123]. The expression of CaSR in inflammatory cells may extend its roles to neuroinflammation [124]. Since soluble Aβ reportedly binds CaSR and leads to neuronal inflammation and cell death [125][126], GSH or other related peptides may be able to exert protective action in the neuronal system through modulating the function of CaSR. Some of the γ-glutamyl peptides that are produced through either the GSH-synthesizing pathway or the GGT-mediated transfer of a γ-glutamyl group may play roles in calcium homeostasis in the neuronal system via modulating CaSR.
References
- Brigelius-Flohé, R.; Flohé, L. Regulatory Phenomena in the Glutathione Peroxidase Superfamily. Antioxid. Redox Signal. 2020, 33, 498–516.
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.
- Cho, C.S.; Lee, S.; Lee, G.T.; Woo, H.A.; Choi, E.J.; Rhee, S.G. Irreversible inactivation of glutathione peroxidase 1 and reversible inactivation of peroxiredoxin II by H2O2 in red blood cells. Antioxid. Redox Signal. 2010, 12, 1235–1246.
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421.
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072.
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331.
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185.
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191.
- Fisher, A.B.; Vasquez-Medina, J.P.; Dodia, C.; Sorokina, E.M.; Tao, J.Q.; Feinstein, S.I. Peroxiredoxin 6 phospholipid hydroperoxidase activity in the repair of peroxidized cell membranes. Redox Biol. 2018, 14, 41–46.
- Cardoso, B.R.; Hare, D.J.; Bush, A.I.; Roberts, B.R. Glutathione peroxidase 4: A new player in neurodegeneration? Mol. Psychiatry 2017, 22, 328–335.
- Fujii, J.; Ito, J.I.; Zhang, X.; Kurahashi, T. Unveiling the roles of the glutathione redox system in vivo by analyzing genetically modified mice. J. Clin. Biochem. Nutr. 2011, 49, 70–78.
- Lillig, C.H.; Berndt, C.; Holmgren, A. Glutaredoxin systems. Biochim. Biophys. Acta 2008, 1780, 1304–1317.
- Brigelius, R.; Muckel, C.; Akerboom, T.P.; Sies, H. Identification and quantitation of glutathione in hepatic protein mixed disulfides and its relationship to glutathione disulfide. Biochem. Pharmacol. 1983, 32, 2529–2534.
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Colombo, R.; Milzani, A. S-glutathionylation in protein redox regulation. Free Radic. Biol. Med. 2007, 43, 883–898.
- Holmgren, A. Hydrogen donor system for Escherichia coli ribonucleoside-diphosphate reductase dependent upon glutathione. Proc. Natl. Acad. Sci. USA 1976, 73, 2275–2279.
- Ogata, F.T.; Branco, V.; Vale, F.F.; Coppo, L. Glutaredoxin: Discovery, redox defense and much more. Redox Biol. 2021, 43, 101975.
- Matsui, R.; Ferran, B.; Oh, A.; Croteau, D.; Shao, D.; Han, J.; Pimentel, D.R.; Bachschmid, M.M. Redox Regulation via Glutaredoxin-1 and Protein S-Glutathionylation. Antioxid. Redox Signal. 2020, 32, 677–700.
- Maher, P. Redox control of neural function: Background, mechanisms, and significance. Antioxid. Redox Signal. 2006, 8, 1941–1970.
- Sabens Liedhegner, E.A.; Gao, X.H.; Mieyal, J.J. Mechanisms of altered redox regulation in neurodegenerative diseases--focus on S--glutathionylation. Antioxid. Redox Signal. 2012, 16, 543–566.
- Kenchappa, R.S.; Diwakar, L.; Boyd, M.R.; Ravindranath, V. Thioltransferase (glutaredoxin) mediates recovery of motor neurons from excitotoxic mitochondrial injury. J. Neurosci. 2002, 22, 8402–8410.
- Diwakar, L.; Kenchappa, R.S.; Annepu, J.; Ravindranath, V. Downregulation of glutaredoxin but not glutathione loss leads to mitochondrial dysfunction in female mice CNS: Implications in excitotoxicity. Neurochem. Int. 2007, 51, 37–46.
- Saeed, U.; Durgadoss, L.; Valli, R.K.; Joshi, D.C.; Joshi, P.G.; Ravindranath, V. Knockdown of cytosolic glutaredoxin 1 leads to loss of mitochondrial membrane potential: Implication in neurodegenerative diseases. PLoS One 2008, 3, e2459.
- Arodin, L.; Miranda-Vizuete, A.; Swoboda, P.; Fernandes, A.P. Protective effects of the thioredoxin and glutaredoxin systems in dopamine-induced cell death. Free Radic. Biol. Med. 2014, 3, 328–336.
- Johnson, W.M.; Golczak, M.; Choe, K.; Curran, P.L.; Miller, O.G.; Yao, C.; Wang, W.; Lin, J.; Milkovic, N.M.; Ray, A.; et al. Regulation of DJ-1 by Glutaredoxin 1 in Vivo: Implications for Parkinson’s Disease. Biochemistry 2016, 55, 4519–4532.
- Romero, J.I.; Hanschmann, E.M.; Gellert, M.; Eitner, S.; Holubiec, M.I.; Blanco-Calvo, E.; Lillig, C.H.; Capani, F. Thioredoxin 1 and glutaredoxin 2 contribute to maintain the phenotype and integrity of neurons following perinatal asphyxia. Biochim. Biophys. Acta 2015, 1850, 1274–1285.
- Karunakaran, S.; Saeed, U.; Ramakrishnan, S.; Koumar, R.C.; Ravindranath, V. Constitutive expression and functional characterization of mitochondrial glutaredoxin (Grx2) in mouse and human brain. Brain Res. 2007, 1185, 8–17.
- Verma, A.; Ray, A.; Bapat, D.; Diwakar, L.; Kommaddi, R.P.; Schneider, B.L.; Hirsch, E.C.; Ravindranath, V. Glutaredoxin 1 Downregulation in the Substantia Nigra Leads to Dopaminergic Degeneration in Mice. Mov. Disord. 2020, 35, 1843–1853.
- Choi, Y.J.; Kim, D.W.; Shin, M.J.; Yeo, H.J.; Yeo, E.J.; Lee, L.R.; Song, Y.; Kim, D.S.; Han, K.H.; Park, J.; et al. PEP-1-GLRX1 Reduces Dopaminergic Neuronal Cell Loss by Modulating MAPK and Apoptosis Signaling in Parkinson’s Disease. Molecules 2021, 26, 3329.
- Lee, D.W.; Kaur, D.; Chinta, S.J.; Rajagopalan, S.; Andersen, J.K. A disruption in iron-sulfur center biogenesis via inhibition of mitochondrial dithiol glutaredoxin 2 may contribute to mitochondrial and cellular iron dysregulation in mammalian glutathione-depleted dopaminergic cells: Implications for Parkinson’s disease. Antioxid. Redox Signal. 2009, 11, 2083–2094.
- Lepka, K.; Volbracht, K.; Bill, E.; Schneider, R.; Rios, N.; Hildebrandt, T.; Ingwersen, J.; Prozorovski, T.; Lillig, C.H.; van Horssen, J.; et al. Iron-sulfur glutaredoxin 2 protects oligodendrocytes against damage induced by nitric oxide release from activated microglia. Glia 2017, 65, 1521–1534.
- Gorelenkova Miller, O.; Behring, J.B.; Siedlak, S.L.; Jiang, S.; Matsui, R.; Bachschmid, M.M.; Zhu, X.; Mieyal, J.J. Upregulation of Glutaredoxin-1 Activates Microglia and Promotes Neurodegeneration: Implications for Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 967–982.
- Johnson, W.M.; Yao, C.; Siedlak, S.L.; Wang, W.; Zhu, X.; Caldwell, G.A.; Wilson-Delfosse, A.L.; Mieyal, J.J.; Chen, S.G. Glutaredoxin deficiency exacerbates neurodegeneration in C. elegans models of Parkinson’s disease. Hum. Mol. Genet. 2015, 24, 1322–1335.
- Kommaddi, R.P.; Tomar, D.S.; Karunakaran, S.; Bapat, D.; Nanguneri, S.; Ray, A.; Schneider, B.L.; Nair, D.; Ravindranath, V. Glutaredoxin1 Diminishes Amyloid Beta-Mediated Oxidation of F-Actin and Reverses Cognitive Deficits in an Alzheimer’s Disease Mouse Model. Antioxid Redox. Signal. 2019, 31, 1321–1338.
- Akterin, S.l; Cowburn, R.F.; Miranda-Vizuete, A.; Jiménez, A.; Bogdanovic, N.; Winblad, B.; Cedazo-Minguez, A. Involvement of glutaredoxin-1 and thioredoxin-1 in beta-amyloid toxicity and Alzheimer’s disease. Cell. Death Differ. 2006, 13, 1454–1465.
- Ferri, A.; Fiorenzo, P.; Nencini, M.; Cozzolino, M.; Pesaresi, M.G.; Valle, C.; Sepe, S.; Moreno, S.; Carrì, M.T. Glutaredoxin 2 prevents aggregation of mutant SOD1 in mitochondria and abolishes its toxicity. Hum. Mol. Genet. 2010, 19, 4529–4542.
- Sau, A.; Pellizzari Tregno, F.; Valentino, F.; Federici, G.; Caccuri, A.M. Glutathione transferases and development of new principles to overcome drug resistance. Arch. Biochem. Biophys. 2010, 500, 116–122.
- Dringen, R.; Brandmann, M.; Hohnholt, M.C.; Blumrich, E.M. Glutathione-Dependent Detoxification Processes in Astrocytes. Neurochem. Res. 2015, 40, 2570–2582.
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88.
- Board, P.G. The use of glutathione transferase-knockout mice as pharmacological and toxicological models. Expert Opin. Drug Metab. Toxicol. 2007, 3, 421–433.
- Henderson, C.J.; Wolf, C.R. Knockout and transgenic mice in glutathione transferase research. Drug Metab. Rev. 2011, 43, 152–164.
- Smeyne, M.; Smeyne, R.J. Glutathione metabolism and Parkinson’s disease. Free Radic. Biol. Med. 2013, 62, 13–25.
- Botta, D.; Shi, S.; White, C.C.; Dabrowski, M.J.; Keener, C.L.; Srinouanprachanh, S.L.; Farin, F.M.; Ware, C.B.; Ladiges, W.C.; Pierce, R.H.; et al. Acetaminophen-induced liver injury is attenuated in male glutamate-cysteine ligase transgenic mice. J. Biol. Chem. 2006, 281, 28865–28875.
- Kanaoka, Y.; Austen, K.F. Roles of cysteinyl leukotrienes and their receptors in immune cell-related functions. Adv. Immunol. 2019, 142, 65–84.
- Gelosa, P.; Colazzo, F.; Tremoli, E.; Sironi, L.; Castiglioni, L. Cysteinyl Leukotrienes as Potential Pharmacological Targets for Cerebral Diseases. Mediators Inflamm. 2017, 2017, 3454212.
- Lam, B.K. Leukotriene C(4) synthase. Prostaglandins Leukot. Essent. Fatty Acids. 2003, 69, 111–116.
- Wang, Y.; Yang, Y.; Zhang, S.; Li, C.; Zhang, L. Modulation of neuroinflammation by cysteinyl leukotriene 1 and 2 receptors: Implications for cerebral ischemia and neurodegenerative diseases. Neurobiol. Aging 2020, 87, 1–10.
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424.
- Jaffrey, S.R.; Erdjument-Bromage, H.; Ferris, C.D.; Tempst, P.; Snyder, S.H. Protein S-nitrosylation: A physiological signal for neuronal nitric oxide. Nat. Cell Biol. 2001, 3, 193–197.
- Anand, P.; Hausladen, A.; Wang, Y.J.; Zhang, G.F.; Stomberski, C.; Brunengraber, H.; Hess, D.T.; Stamler, J.S. Identification of S-nitroso-CoA reductases that regulate protein S-nitrosylation. Proc. Natl. Acad. Sci. USA 2014, 111, 18572–18577.
- Sircar, E.; Rai, S.R.; Wilson, M.A.; Schlossmacher, M.G.; Sengupta, R. Neurodegeneration: Impact of S-nitrosylated Parkin, DJ-1 and PINK1 on the pathogenesis of Parkinson’s disease. Arch. Biochem. Biophys. 2021, 704, 108869.
- Seth, D.; Hess, D.T.; Hausladen, A.; Wang, L.; Wang, Y.J.; Stamler, J.S. A Multiplex Enzymatic Machinery for Cellular Protein S-nitrosylation. Mol. Cell 2018, 69, 451–464.e6.
- Stomberski, C.T.; Hess, D.T.; Stamler, J.S. Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling. Antioxid. Redox Signal. 2019, 30, 1331–1351.
- Ballatori, N.; Krance, S.M.; Marchan, R.; Hammond, C.L. Plasma membrane glutathione transporters and their roles in cell physiology and pathophysiology. Mol. Aspects Med. 2009, 30, 13–28.
- Benhar, M.; Forrester, M.T.; Stamler, J.S. Protein denitrosylation: Enzymatic mechanisms and cellular functions. Nat. Rev. Mol. Cell Biol. 2009, 10, 721–732.
- Jensen, D.E.; Belka, G.K.; Du Bois, G.C. S-Nitrosoglutathione is a substrate for rat alcohol dehydrogenase class III isoenzyme. Biochem. J. 1998, 331, 659–668.
- Liu, L.; Hausladen, A.; Zeng, M.; Que, L.; Heitman, J.; Stamler, J.S. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature 2001, 410, 490–494.
- Bateman, R.L.; Rauh, D.; Tavshanjian, B.; Shokat, K.M. Human carbonyl reductase 1 is an S-nitrosoglutathione reductase. J. Biol. Chem. 2008, 283, 35756–35762.
- Rizza, S.; Di Leo, L.; Pecorari, C.; Giglio, P.; Faienza, F.; Montagna, C.; Maiani, E.; Puglia, M.; Bosisio, F.M.; Petersen, T.S.; et al. GSNOR deficiency promotes tumor growth via FAK1 S-nitrosylation. Cell Rep. 2023, 42, 111997.
- Nakamura, T.; Oh, C.K.; Zhang, X.; Lipton, S.A. Protein S-nitrosylation and oxidation contribute to protein misfolding in neurodegeneration. Free Radic. Biol. Med. 2021, 172, 562–577.
- Vrettou, S.; Wirth, B. S-Glutathionylation and S-Nitrosylation in Mitochondria: Focus on Homeostasis and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 15849.
- Jyotsana, N.; Ta, K.T.; DelGiorno, K.E. The Role of Cystine/Glutamate Antiporter SLC7A11/xCT in the Pathophysiology of Cancer. Front. Oncol. 2022, 12, 858462.
- Reichert, C.O.; de Freitas, F.A.; Sampaio-Silva, J.; Rokita-Rosa, L.; Barros, P.L.; Levy, D.; Bydlowski, S.P. Ferroptosis Mechanisms Involved in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8765.
- Palaniyappan, L.; Park, M.T.M.; Jeon, P.; Limongi, R.; Yang, K.; Sawa, A.; Théberge, J. Is There a Glutathione Centered Redox Dysregulation Subtype of Schizophrenia? Antioxidants 2021, 10, 1703.
- Peeples, E.S.; Genaro-Mattos, T.C. Ferroptosis: A Promising Therapeutic Target for Neonatal Hypoxic-Ischemic Brain Injury. Int. J. Mol. Sci. 2022, 23, 7420.
- Gleitze, S.; Paula-Lima, A.; Núñez, M.T.; Hidalgo, C. The calcium-iron connection in ferroptosis-mediated neuronal death. Free Radic. Biol. Med. 2021, 175, 28–41.
- Chen, K.; Jiang, X.; Wu, M.; Cao, X.; Bao, W.; Zhu, L.Q. Ferroptosis, a Potential Therapeutic Target in Alzheimer’s Disease. Front. Cell Dev. Biol. 2021, 9, 704298.
- Jakaria, M.; Belaidi, A.A.; Bush, A.I.; Ayton, S. Ferroptosis as a mechanism of neurodegeneration in Alzheimer’s disease. J. Neurochem. 2021, 159, 804–825.
- Crapper-McLachlan., D.R.; Dalton, A.J.; Kruck, T.P.; Bell, M.Y.; Smith, W.L.; Kalow, W.; Andrews, D.F. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 1991, 337, 1304–1308.
- Dysken, M.W.; Sano, M.; Asthana, S.; Vertrees, J.E.; Pallaki, M.; Llorente, M.; Love, S.; Schellenberg, G.D.; McCarten, J.R.; Malphurs, J.; et al. Effect of vitamin E and memantine on functional decline in Alzheimer disease: The TEAM-AD VA cooperative randomized trial. JAMA 2014, 311, 33–44.
- Da Costa, M.; Bernardi, J.; Costa, L.; Fiuza, T.; Brandão, R.; Ribeiro, M.F.; Amaral, J.D.; Rodrigues, C.M.P.; Pereira, M.E. N-acetylcysteine treatment attenuates the cognitive impairment and synaptic plasticity loss induced by streptozotocin. Chem. Biol. Interact. 2017, 272, 37–46.
- More, J.; Galusso, N.; Veloso, P.; Montecinos, L.; Finkelstein, J.P.; Sanchez, G.; Bull, R.; Valdés, J.L.; Hidalgo, C.; Paula-Lima, A. N-Acetylcysteine Prevents the Spatial Memory Deficits and the Redox-Dependent RyR2 Decrease Displayed by an Alzheimer’s Disease Rat Model. Front. Aging Neurosci. 2018, 10, 399.
- Stapper, Z.A.; Jahn, T.R. Changes in Glutathione Redox Potential Are Linked to Aβ42-Induced Neurotoxicity. Cell Rep. 2018, 24, 1696–1703.
- Guiney, S.J.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem. Int. 2017, 104, 34–48.
- Lin, K.J.; Chen, S.D.; Lin, K.L.; Liou, C.W.; Lan, M.Y.; Chuang, Y.C.; Wang, P.W.; Lee, J.J.; Wang, F.S.; Lin, H.Y.; et al. Iron Brain Menace: The Involvement of Ferroptosis in Parkinson Disease. Cells 2022, 11, 3829.
- Thapa, K.; Khan, H.; Kanojia, N.; Singh, T.G.; Kaur, A.; Kaur, G. Therapeutic Insights on Ferroptosis in Parkinson’s disease. Eur. J. Pharmacol. 2022, 930, 175133.
- Bisaglia, M.; Soriano, M.E.; Arduini, I.; Mammi, S.; Bubacco, L. Molecular characterization of dopamine-derived quinones reactivity toward NADH and glutathione: Implications for mitochondrial dysfunction in Parkinson disease. Biochim. Biophys. Acta 2010, 1802, 699–706.
- Segura-Aguilar, J.; Paris, I.; Muñoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915.
- Asanuma, M.; Miyazaki, I. Glutathione and Related Molecules in Parkinsonism. Int. J. Mol. Sci. 2021, 22, 8689.
- Cole, S.P.; Deeley, R.G. Transport of glutathione and glutathione conjugates by MRP1. Trends Pharmacol. Sci. 2006, 27, 438–446.
- Badillo-Ramírez, I.; Saniger, J.M.; Rivas-Arancibia, S. 5-S-cysteinyl-dopamine, a neurotoxic endogenous metabolite of dopamine: Implications for Parkinson’s disease. Neurochem. Int. 2019, 129, 104514.
- Stern, S.T.; Bruno, M.K.; Hennig, G.E.; Horton, R.A.; Roberts, J.C.; Cohen, S.D. Contribution of acetaminophen-cysteine to acetaminophen nephrotoxicity in CD-1 mice: I. Enhancement of acetaminophen nephrotoxicity by acetaminophen-cysteine. Toxicol. Appl. Pharmacol. 2005, 202, 151–159.
- Kim, K. Glutathione in the Nervous System as a Potential Therapeutic Target to Control the Development and Progression of Amyotrophic Lateral Sclerosis. Antioxidants 2021, 10, 1011.
- Valle, C.; Carrì, M.T. Cysteine Modifications in the Pathogenesis of ALS. Front. Mol. Neurosci. 2017, 10, 5.
- Pang, Y.; Liu, X.; Wang, X.; Shi, X.; Ma, L.; Zhang, Y.; Zhou, T.; Zhao, C.; Zhang, X.; Fan, B.; et al. Edaravone Modulates Neuronal GPX4/ACSL4/5-LOX to Promote Recovery After Spinal Cord Injury. Front. Cell Dev. Biol. 2022, 10, 849854.
- Watanabe, K.; Tanaka, M.; Yuki, S.; Hirai, M.; Yamamoto, Y. How is edaravone effective against acute ischemic stroke and amyotrophic lateral sclerosis? J. Clin. Biochem. Nutr. 2018, 62, 20–38.
- Spasić, S.; Nikolić-Kokić, A.; Miletić, S.; Oreščanin-Dušić, Z.; Spasić, M.B.; Blagojević, D.; Stević, Z. Edaravone May Prevent Ferroptosis in ALS. Curr. Drug Targets 2020, 21, 776–780.
- Stipanuk, M.H.; Ueki, I.; Dominy, J.E., Jr.; Simmons, C.R.; Hirschberger, L.L. Cysteine dioxygenase: A robust system for regulation of cellular cysteine levels. Amino Acids 2009, 37, 55–63.
- Wright, C.E.; Tallan, H.H.; Lin, Y.Y.; Gaull, G.E. Taurine: Biological update. Annu. Rev. Biochem. 1986, 55, 427–453.
- Chen, Q.; Konrad, C.; Sandhu, D.; Roychoudhury, D.; Schwartz, B.I.; Cheng, R.R.; Bredvik, K.; Kawamata, H.; Calder, E.L.; Studer, L.; et al. Accelerated transsulfuration metabolically defines a discrete subclass of amyotrophic lateral sclerosis patients. Neurobiol. Dis. 2020, 144, 105025.
- Varga, V.; Török, K.; Feuer, L.; Gulyás, J.; Somogyi, J. γ-Glutamyltransferase in the brain and its role in formation of γ-L-glutamyl-taurine. Prog. Clin. Biol. Res. 1985, 179, 115–125.
- Kobayashi, S.; Tokairin, Y.; Miyakoshi, T.; Saito, T.; Nagaoka, T.; Ikeda, Y.; Fujii, J.; Konno, H. Quantitative analysis of γ-glutamylpeptides by liquid chromatography-mass spectrometry and application for γ-glutamyltransferase assays. Anal. Biochem. 2019, 578, 13–22.
- Kobayashi, S.; Ikeda, Y.; Shigeno, Y.; Konno, H.; Fujii, J. γ-Glutamylcysteine synthetase and γ-glutamyl transferase as differential enzymatic sources of γ-glutamylpeptides in mice. Amino Acids 2020, 52, 555–566.
- Varga, V.; Janáky, R.; Marnela, K.M.; Saransaari, P.; Oja, S.S. Interactions of gamma-L-glutamyltaurine with excitatory aminoacidergic neurotransmission. Neurochem. Res. 1994, 19, 243–248.
- Uemura, S.; Ienaga, K.; Higashiura, K.; Kimura, H. Gamma-glutamyltaurine has potent and long-lasting antiepileptic action as demonstrated by intra-amygdaloid injection in amygdala-kindled rats. Brain Res. 1992, 594, 347–350.
- Bittner, S.; Win, T.; Gupta, R. γ-L-glutamyltaurine. Amino Acids 2005, 28, 343–356.
- Fujii, J.; Homma, T.; Kobayashi, S. Ferroptosis caused by cysteine insufficiency and oxidative insult. Free Radic. Res. 2020, 54, 969–980.
- Griffith, O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med. 1999, 27, 922–935.
- Orlowski, M.; Wilk, S. Intermediates of the gamma-glutamyl cycle in mouse tissues. Influence of administration of amino acids on pyrrolidone carboxylate and gamma-glutamyl amino acids. Eur. J. Biochem. 1975, 53, 581–590.
- Soga, T.; Baran, R.; Suematsu, M.; Ueno, Y.; Ikeda, S.; Sakurakawa, T.; Kakazu, Y.; Ishikawa, T.; Robert, M.; Nishioka, T.; et al. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J. Biol. Chem. 2006, 281, 16768–16776.
- Kobayashi, S.; Lee, J.; Takao, T.; Fujii, J. Increased ophthalmic acid production is supported by amino acid catabolism under fasting conditions in mice. Biochem. Biophys. Res. Commun. 2017, 491, 649–655.
- Soga, T.; Sugimoto, M.; Honma, M.; Mori, M.; Igarashi, K.; Kashikura, K.; Ikeda, S.; Hirayama, A.; Yamamoto, T.; Yoshida, H.; et al. Serum metabolomics reveals γ-glutamyl dipeptides as biomarkers for discrimination among different forms of liver disease. J. Hepatol. 2011, 55, 896–905.
- Deepmala; Slattery, J.; Kumar, N.; Delhey, L.; Berk, M.; Dean, O.; Spielholz, C.; Frye, R. Clinical trials of N-acetylcysteine in psychiatry and neurology: A systematic review. Neurosci. Biobehav. Rev. 2015, 55, 294–321.
- Kang, Y.P.; Mockabee-Macias, A.; Jiang, C.; Falzone, A.; Prieto-Farigua, N.; Stone, E.; Harris, I.S.; DeNicola, G.M. Non-canonical Glutamate-Cysteine Ligase Activity Protects against Ferroptosis. Cell Metab. 2021, 33, 174–189.e7.
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.
- Homma, T.; Kobayashi, S.; Fujii, J. Methionine Deprivation Reveals the Pivotal Roles of Cell Cycle Progression in Ferroptosis That Is Induced by Cysteine Starvation. Cells 2022, 11, 1603.
- Maclean, K.N.; Jiang, H.; Aivazidis, S.; Kim, E.; Shearn, C.T.; Harris, P.S.; Petersen, D.R.; Allen, R.H.; Stabler, S.P.; Roede, J.R. Taurine treatment prevents derangement of the hepatic γ-glutamyl cycle and methylglyoxal metabolism in a mouse model of classical homocystinuria: Regulatory crosstalk between thiol and sulfinic acid metabolism. FASEB J. 2018, 32, 1265–1280.
- Li, H.Q.; Xia, S.N.; Xu, S.Y.; Liu, P.Y.; Gu, Y.; Bao, X.Y.; Xu, Y.; Cao, X. γ-Glutamylcysteine Alleviates Ischemic Stroke-Induced Neuronal Apoptosis by Inhibiting ROS-Mediated Endoplasmic Reticulum Stress. Oxid. Med. Cell Longev. 2021, 2021, 2961079.
- Zhang, H.; Kovacs-Nolan, J.; Kodera, T.; Eto, Y.; Mine, Y. γ-Glutamyl cysteine and γ-glutamyl valine inhibit TNF-α signaling in intestinal epithelial cells and reduce inflammation in a mouse model of colitis via allosteric activation of the calcium-sensing receptor. Biochim. Biophys. Acta 2015, 1852, 792–804.
- Hewson, C.K.; Capraro, A.; Wong, S.L.; Pandzic, E.; Zhong, L.; Fernando, B.S.M.; Awatade, N.T.; Hart-Smith, G.; Whan, R.M.; Thomas, S.R.; et al. Novel Antioxidant Therapy with the Immediate Precursor to Glutathione, γ-Glutamylcysteine (GGC), Ameliorates LPS-Induced Cellular Stress in In Vitro 3D-Differentiated Airway Model from Primary Cystic Fibrosis Human Bronchial Cells. Antioxidants 2020, 9, 1204.
- Bi, A.; Wang, Y.; Chen, L.; Yin, Z.; Luo, L. γ-Glutamylcysteine attenuates amyloid-β oligomers-induced neuroinflammation in microglia via blocking NF-κB signaling pathway. Chem. Biol. Interact. 2022, 363, 110019.
- Zhou, J.; Yan, X.; Bi, X.; Lu, S.; Liu, X.; Yang, C.; Shi, Y.; Luo, L.; Yin, Z. γ-Glutamylcysteine rescues mice from TNBS-driven inflammatory bowel disease through regulating macrophages polarization. Inflamm. Res. 2023, 72, 603–621.
- Liu, Y.; Chen, Z.; Li, B.; Yao, H.; Zarka, M.; Welch, J.; Sachdev, P.; Bridge, W.; Braidy, N. Supplementation with γ-glutamylcysteine (γ-GC) lessens oxidative stress, brain inflammation and amyloid pathology and improves spatial memory in a murine model of AD. Neurochem. Int. 2021, 144, 104931.
- Zhang, R.; Lei, J.; Chen, L.; Wang, Y.; Yang, G.; Yin, Z.; Luo, L. γ-Glutamylcysteine Exerts Neuroprotection Effects against Cerebral Ischemia/Reperfusion Injury through Inhibiting Lipid Peroxidation and Ferroptosis. Antioxidants 2022, 11, 1653.
- Ferguson, G.; Bridge, W. Glutamate cysteine ligase and the age-related decline in cellular glutathione: The therapeutic potential of γ-glutamylcysteine. Arch. Biochem. Biophys. 2016, 593, 12–23.
- Zarka, M.H.; Bridge, W.J. Oral administration of γ-glutamylcysteine increases intracellular glutathione levels above homeostasis in a randomised human trial pilot study. Redox Biol. 2017, 11, 631–636.
- Yang, Y.; Li, L.; Hang, Q.; Fang, Y.; Dong, X.; Cao, P.; Yin, Z.; Luo, L. γ-glutamylcysteine exhibits anti-inflammatory effects by increasing cellular glutathione level. Redox Biol. 2019, 20, 157–166.
- Quintana-Cabrera, R.; Fernandez-Fernandez, S.; Bobo-Jimenez, V.; Escobar, J.; Sastre, J.; Almeida, A.; Bolaños, J.P. γ-Glutamylcysteine detoxifies reactive oxygen species by acting as glutathione peroxidase-1 cofactor. Nat. Commun. 2012, 3, 718.
- Wang, M.; Yao, Y.; Kuang, D.; Hampson, D.R. Activation of family C G-protein-coupled receptors by the tripeptide glutathione. J. Biol. Chem. 2006, 281, 8864–8870.
- Goralski, T.; Ram, J.L. Extracellular Calcium Receptor as a Target for Glutathione and Its Derivatives. Int. J. Mol. Sci. 2022, 23, 717.
- Bandyopadhyay, S.; Tfelt-Hansen, J.; Chattopadhyay, N. Diverse roles of extracellular calcium-sensing receptor in the central nervous system. J. Neurosci Res. 2010, 88, 2073–2082.
- Brennan, S.C.; Davies, T.S.; Schepelmann, M.; Riccardi, D. Emerging roles of the extracellular calcium-sensing receptor in nutrient sensing: Control of taste modulation and intestinal hormone secretion. Br. J. Nutr. 2014, 111, S16–S22.
- Guha, S.; Majumder, K. J Comprehensive Review of γ-Glutamyl Peptides (γ-GPs) and Their Effect on Inflammation Concerning Cardiovascular Health. Agric. Food Chem. 2022, 70, 7851–7870.
- Giudice, M.L.; Mihalik, B.; Dinnyés, A.; Kobolák, J. The Nervous System Relevance of the Calcium Sensing Receptor in Health and Disease. Molecules 2019, 24, 2546.
- Iamartino, L.; Brandi, M.L. The calcium-sensing receptor in inflammation: Recent updates. Front. Physiol. 2022, 13, 1059369.
- Chiarini, A.; Armato, U.; Gardenal, E.; Gui, L.; Dal Prà, I. Amyloid β-Exposed Human Astrocytes Overproduce Phospho-Tau and Overrelease It within Exosomes, Effects Suppressed by Calcilytic NPS 2143-Further Implications for Alzheimer’s Therapy. Front. Neurosci. 2017, 11, 217.
- Dal Prà, I.; Armato, U.; Chiarini, A. Family C G-Protein-Coupled Receptors in Alzheimer’s Disease and Therapeutic Implications. Front. Pharmacol. 2019, 10, 1282.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
943
Revisions:
3 times
(View History)
Update Date:
16 May 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No