Methionine (Met) and cysteine (Cys) are the only sulfur-containing amino acids, and Cys can be synthesized through the transsulfuration reaction associated with Met metabolism. Met is metabolized to S-adenosylmethionine (SAM), which provides either the carbon backbone for polyamines or methyl groups for some other compounds, including DNA . Meanwhile, Cys comes from extracellular sources or is the product of the transsulfuration reaction and then becomes the precursor for sulfur-containing components or mediators. These include glutathione (GSH), taurine, coenzyme A, hydrogen sulfide, iron–sulfur [Fe–S] cluster, and persulfides.
- xCT
- glutathione peroxidase
- glutaredoxin
- γ-glutamyl transferase
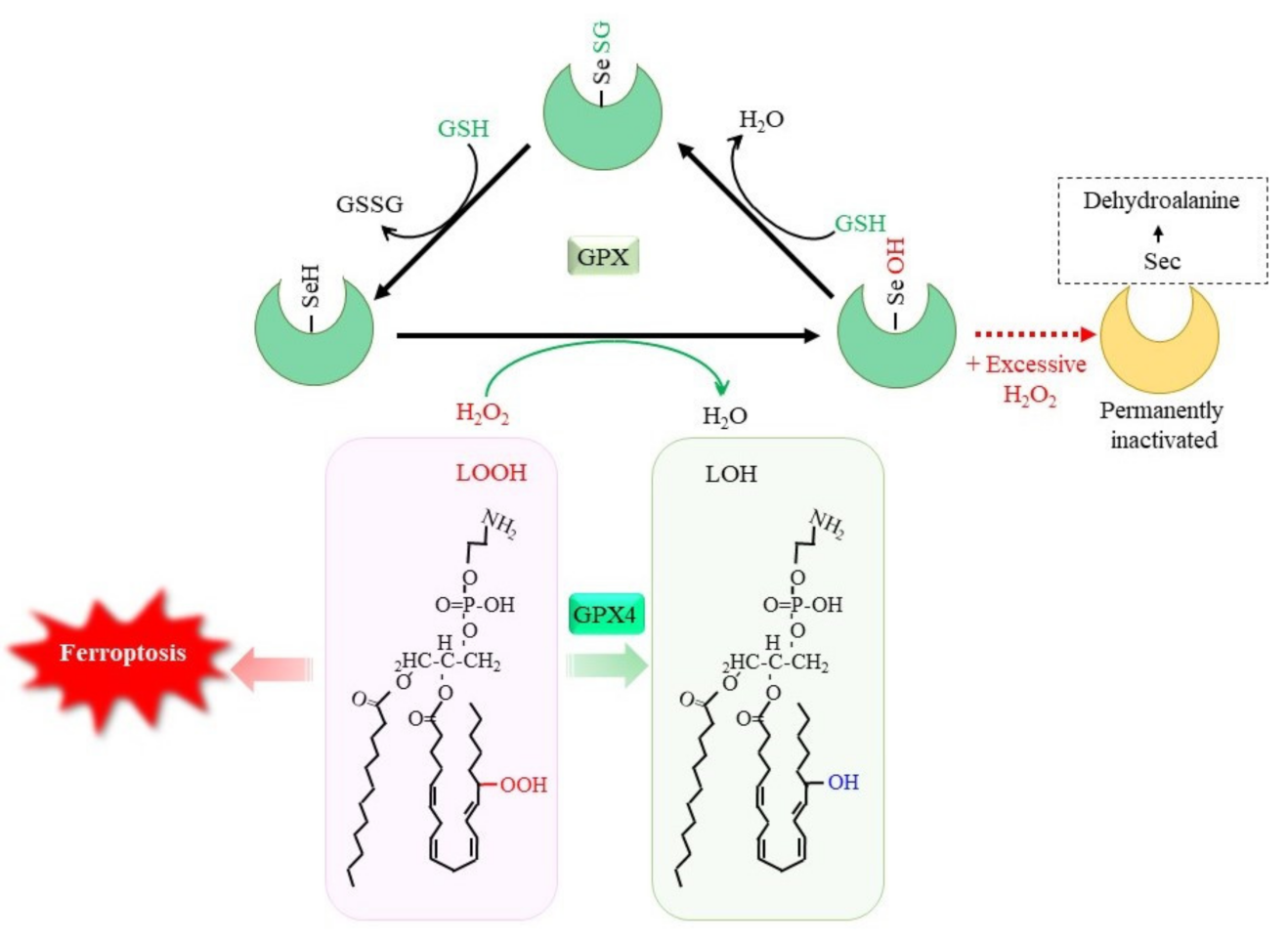
1. Reductive Detoxification of Peroxides via Glutathione Peroxidases
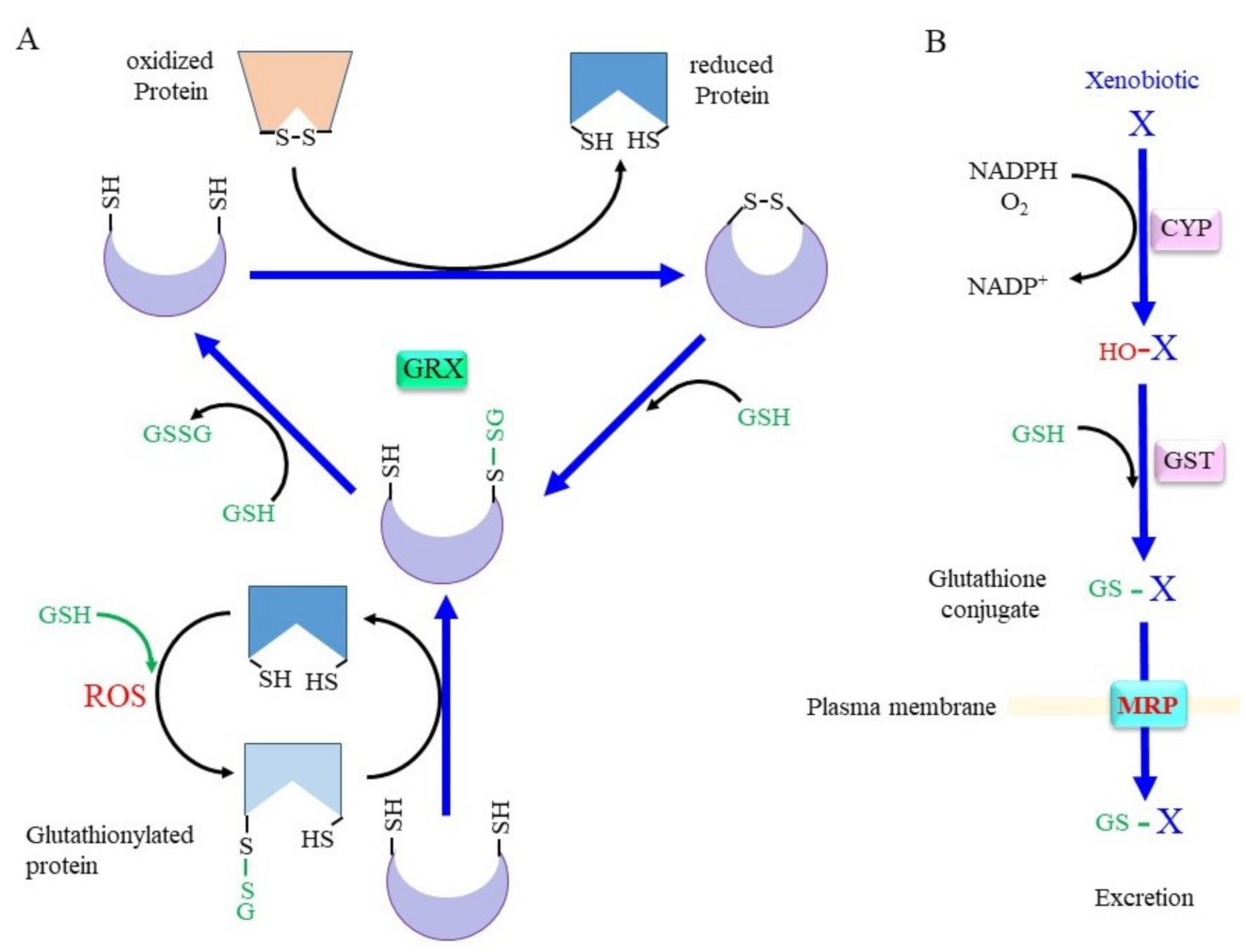
2. Rescuing Proteins from Oxidative Modification through Glutaredoxin
3. Glutathione Conjugation in the Detoxification of Xenobiotics and in the Production of Bioreactive Compounds
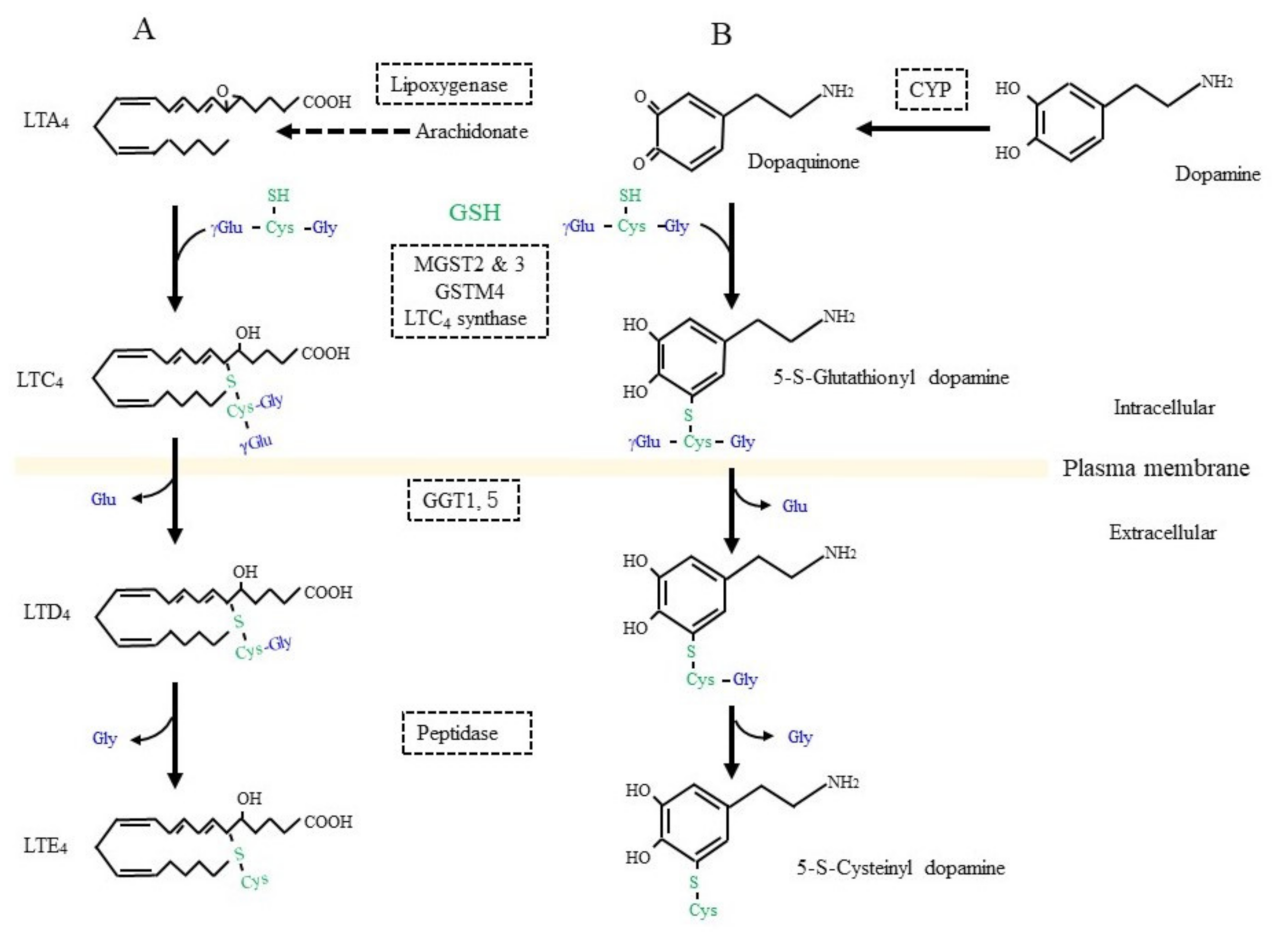
4. Glutathione S-Conjugates, and S-Nitrosoglutathione (GSNO) in Nitric Oxide Signal Transduction
5. Glutathione Status Associated with Neuronal Diseases
6. Production of a Variety of γ-Glutamyl Peptides by Means of γ-Glutamyl–Cysteine Synthetase and γ-Glutamyl Transferase
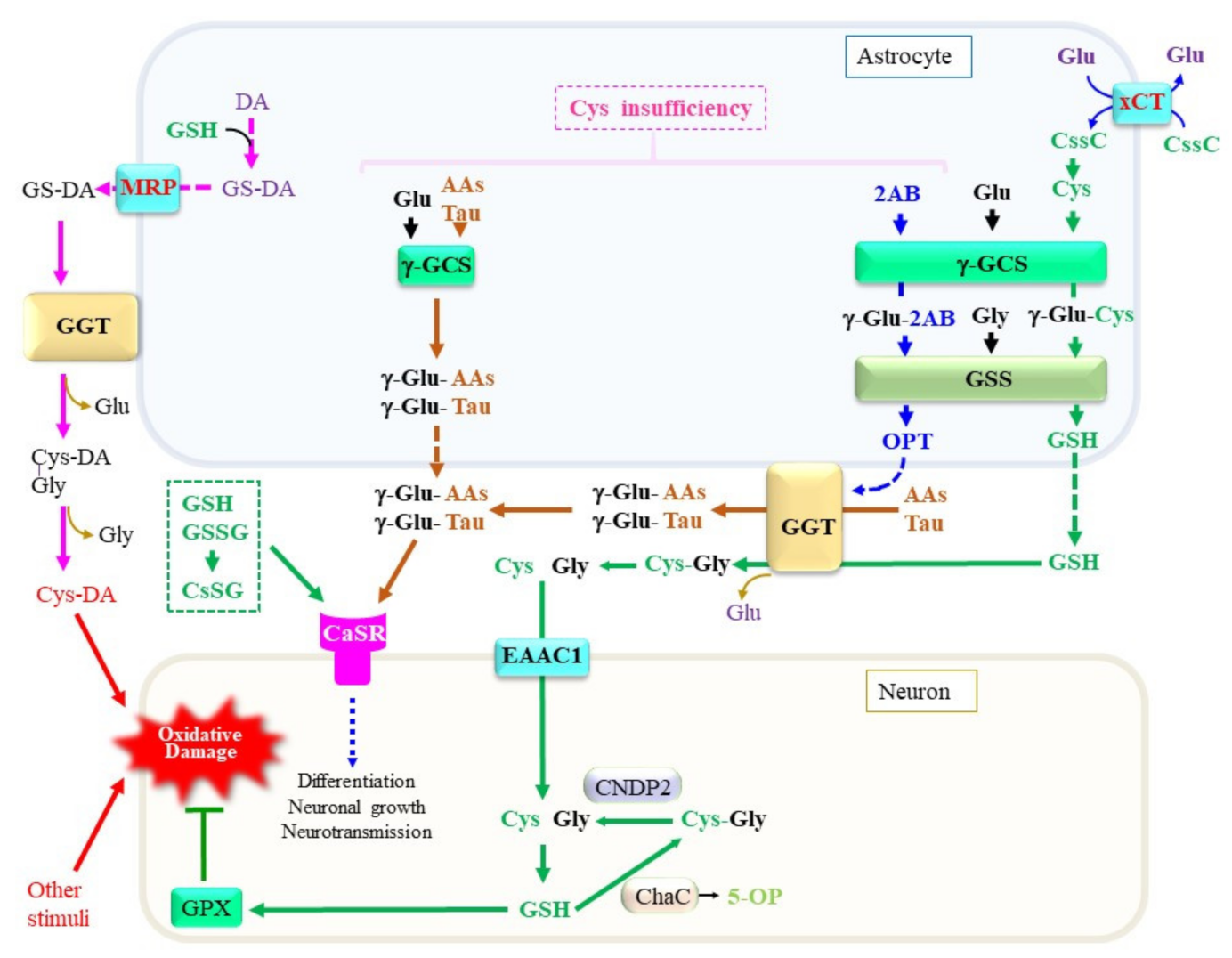
7. Calcium-Sensing Receptor as a Target of γ-Glutamyl Peptides
This entry is adapted from the peer-reviewed paper 10.3390/ijms24098044
References
- Brigelius-Flohé, R.; Flohé, L. Regulatory Phenomena in the Glutathione Peroxidase Superfamily. Antioxid. Redox Signal. 2020, 33, 498–516.
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.
- Cho, C.S.; Lee, S.; Lee, G.T.; Woo, H.A.; Choi, E.J.; Rhee, S.G. Irreversible inactivation of glutathione peroxidase 1 and reversible inactivation of peroxiredoxin II by H2O2 in red blood cells. Antioxid. Redox Signal. 2010, 12, 1235–1246.
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421.
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072.
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331.
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185.
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191.
- Fisher, A.B.; Vasquez-Medina, J.P.; Dodia, C.; Sorokina, E.M.; Tao, J.Q.; Feinstein, S.I. Peroxiredoxin 6 phospholipid hydroperoxidase activity in the repair of peroxidized cell membranes. Redox Biol. 2018, 14, 41–46.
- Cardoso, B.R.; Hare, D.J.; Bush, A.I.; Roberts, B.R. Glutathione peroxidase 4: A new player in neurodegeneration? Mol. Psychiatry 2017, 22, 328–335.
- Fujii, J.; Ito, J.I.; Zhang, X.; Kurahashi, T. Unveiling the roles of the glutathione redox system in vivo by analyzing genetically modified mice. J. Clin. Biochem. Nutr. 2011, 49, 70–78.
- Lillig, C.H.; Berndt, C.; Holmgren, A. Glutaredoxin systems. Biochim. Biophys. Acta 2008, 1780, 1304–1317.
- Brigelius, R.; Muckel, C.; Akerboom, T.P.; Sies, H. Identification and quantitation of glutathione in hepatic protein mixed disulfides and its relationship to glutathione disulfide. Biochem. Pharmacol. 1983, 32, 2529–2534.
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Colombo, R.; Milzani, A. S-glutathionylation in protein redox regulation. Free Radic. Biol. Med. 2007, 43, 883–898.
- Holmgren, A. Hydrogen donor system for Escherichia coli ribonucleoside-diphosphate reductase dependent upon glutathione. Proc. Natl. Acad. Sci. USA 1976, 73, 2275–2279.
- Ogata, F.T.; Branco, V.; Vale, F.F.; Coppo, L. Glutaredoxin: Discovery, redox defense and much more. Redox Biol. 2021, 43, 101975.
- Matsui, R.; Ferran, B.; Oh, A.; Croteau, D.; Shao, D.; Han, J.; Pimentel, D.R.; Bachschmid, M.M. Redox Regulation via Glutaredoxin-1 and Protein S-Glutathionylation. Antioxid. Redox Signal. 2020, 32, 677–700.
- Maher, P. Redox control of neural function: Background, mechanisms, and significance. Antioxid. Redox Signal. 2006, 8, 1941–1970.
- Sabens Liedhegner, E.A.; Gao, X.H.; Mieyal, J.J. Mechanisms of altered redox regulation in neurodegenerative diseases--focus on S--glutathionylation. Antioxid. Redox Signal. 2012, 16, 543–566.
- Kenchappa, R.S.; Diwakar, L.; Boyd, M.R.; Ravindranath, V. Thioltransferase (glutaredoxin) mediates recovery of motor neurons from excitotoxic mitochondrial injury. J. Neurosci. 2002, 22, 8402–8410.
- Diwakar, L.; Kenchappa, R.S.; Annepu, J.; Ravindranath, V. Downregulation of glutaredoxin but not glutathione loss leads to mitochondrial dysfunction in female mice CNS: Implications in excitotoxicity. Neurochem. Int. 2007, 51, 37–46.
- Saeed, U.; Durgadoss, L.; Valli, R.K.; Joshi, D.C.; Joshi, P.G.; Ravindranath, V. Knockdown of cytosolic glutaredoxin 1 leads to loss of mitochondrial membrane potential: Implication in neurodegenerative diseases. PLoS One 2008, 3, e2459.
- Arodin, L.; Miranda-Vizuete, A.; Swoboda, P.; Fernandes, A.P. Protective effects of the thioredoxin and glutaredoxin systems in dopamine-induced cell death. Free Radic. Biol. Med. 2014, 3, 328–336.
- Johnson, W.M.; Golczak, M.; Choe, K.; Curran, P.L.; Miller, O.G.; Yao, C.; Wang, W.; Lin, J.; Milkovic, N.M.; Ray, A.; et al. Regulation of DJ-1 by Glutaredoxin 1 in Vivo: Implications for Parkinson’s Disease. Biochemistry 2016, 55, 4519–4532.
- Romero, J.I.; Hanschmann, E.M.; Gellert, M.; Eitner, S.; Holubiec, M.I.; Blanco-Calvo, E.; Lillig, C.H.; Capani, F. Thioredoxin 1 and glutaredoxin 2 contribute to maintain the phenotype and integrity of neurons following perinatal asphyxia. Biochim. Biophys. Acta 2015, 1850, 1274–1285.
- Karunakaran, S.; Saeed, U.; Ramakrishnan, S.; Koumar, R.C.; Ravindranath, V. Constitutive expression and functional characterization of mitochondrial glutaredoxin (Grx2) in mouse and human brain. Brain Res. 2007, 1185, 8–17.
- Verma, A.; Ray, A.; Bapat, D.; Diwakar, L.; Kommaddi, R.P.; Schneider, B.L.; Hirsch, E.C.; Ravindranath, V. Glutaredoxin 1 Downregulation in the Substantia Nigra Leads to Dopaminergic Degeneration in Mice. Mov. Disord. 2020, 35, 1843–1853.
- Choi, Y.J.; Kim, D.W.; Shin, M.J.; Yeo, H.J.; Yeo, E.J.; Lee, L.R.; Song, Y.; Kim, D.S.; Han, K.H.; Park, J.; et al. PEP-1-GLRX1 Reduces Dopaminergic Neuronal Cell Loss by Modulating MAPK and Apoptosis Signaling in Parkinson’s Disease. Molecules 2021, 26, 3329.
- Lee, D.W.; Kaur, D.; Chinta, S.J.; Rajagopalan, S.; Andersen, J.K. A disruption in iron-sulfur center biogenesis via inhibition of mitochondrial dithiol glutaredoxin 2 may contribute to mitochondrial and cellular iron dysregulation in mammalian glutathione-depleted dopaminergic cells: Implications for Parkinson’s disease. Antioxid. Redox Signal. 2009, 11, 2083–2094.
- Lepka, K.; Volbracht, K.; Bill, E.; Schneider, R.; Rios, N.; Hildebrandt, T.; Ingwersen, J.; Prozorovski, T.; Lillig, C.H.; van Horssen, J.; et al. Iron-sulfur glutaredoxin 2 protects oligodendrocytes against damage induced by nitric oxide release from activated microglia. Glia 2017, 65, 1521–1534.
- Gorelenkova Miller, O.; Behring, J.B.; Siedlak, S.L.; Jiang, S.; Matsui, R.; Bachschmid, M.M.; Zhu, X.; Mieyal, J.J. Upregulation of Glutaredoxin-1 Activates Microglia and Promotes Neurodegeneration: Implications for Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 967–982.
- Johnson, W.M.; Yao, C.; Siedlak, S.L.; Wang, W.; Zhu, X.; Caldwell, G.A.; Wilson-Delfosse, A.L.; Mieyal, J.J.; Chen, S.G. Glutaredoxin deficiency exacerbates neurodegeneration in C. elegans models of Parkinson’s disease. Hum. Mol. Genet. 2015, 24, 1322–1335.
- Kommaddi, R.P.; Tomar, D.S.; Karunakaran, S.; Bapat, D.; Nanguneri, S.; Ray, A.; Schneider, B.L.; Nair, D.; Ravindranath, V. Glutaredoxin1 Diminishes Amyloid Beta-Mediated Oxidation of F-Actin and Reverses Cognitive Deficits in an Alzheimer’s Disease Mouse Model. Antioxid Redox. Signal. 2019, 31, 1321–1338.
- Akterin, S.l; Cowburn, R.F.; Miranda-Vizuete, A.; Jiménez, A.; Bogdanovic, N.; Winblad, B.; Cedazo-Minguez, A. Involvement of glutaredoxin-1 and thioredoxin-1 in beta-amyloid toxicity and Alzheimer’s disease. Cell. Death Differ. 2006, 13, 1454–1465.
- Ferri, A.; Fiorenzo, P.; Nencini, M.; Cozzolino, M.; Pesaresi, M.G.; Valle, C.; Sepe, S.; Moreno, S.; Carrì, M.T. Glutaredoxin 2 prevents aggregation of mutant SOD1 in mitochondria and abolishes its toxicity. Hum. Mol. Genet. 2010, 19, 4529–4542.
- Sau, A.; Pellizzari Tregno, F.; Valentino, F.; Federici, G.; Caccuri, A.M. Glutathione transferases and development of new principles to overcome drug resistance. Arch. Biochem. Biophys. 2010, 500, 116–122.
- Dringen, R.; Brandmann, M.; Hohnholt, M.C.; Blumrich, E.M. Glutathione-Dependent Detoxification Processes in Astrocytes. Neurochem. Res. 2015, 40, 2570–2582.
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88.
- Board, P.G. The use of glutathione transferase-knockout mice as pharmacological and toxicological models. Expert Opin. Drug Metab. Toxicol. 2007, 3, 421–433.
- Henderson, C.J.; Wolf, C.R. Knockout and transgenic mice in glutathione transferase research. Drug Metab. Rev. 2011, 43, 152–164.
- Smeyne, M.; Smeyne, R.J. Glutathione metabolism and Parkinson’s disease. Free Radic. Biol. Med. 2013, 62, 13–25.
- Botta, D.; Shi, S.; White, C.C.; Dabrowski, M.J.; Keener, C.L.; Srinouanprachanh, S.L.; Farin, F.M.; Ware, C.B.; Ladiges, W.C.; Pierce, R.H.; et al. Acetaminophen-induced liver injury is attenuated in male glutamate-cysteine ligase transgenic mice. J. Biol. Chem. 2006, 281, 28865–28875.
- Kanaoka, Y.; Austen, K.F. Roles of cysteinyl leukotrienes and their receptors in immune cell-related functions. Adv. Immunol. 2019, 142, 65–84.
- Gelosa, P.; Colazzo, F.; Tremoli, E.; Sironi, L.; Castiglioni, L. Cysteinyl Leukotrienes as Potential Pharmacological Targets for Cerebral Diseases. Mediators Inflamm. 2017, 2017, 3454212.
- Lam, B.K. Leukotriene C(4) synthase. Prostaglandins Leukot. Essent. Fatty Acids. 2003, 69, 111–116.
- Wang, Y.; Yang, Y.; Zhang, S.; Li, C.; Zhang, L. Modulation of neuroinflammation by cysteinyl leukotriene 1 and 2 receptors: Implications for cerebral ischemia and neurodegenerative diseases. Neurobiol. Aging 2020, 87, 1–10.
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424.
- Jaffrey, S.R.; Erdjument-Bromage, H.; Ferris, C.D.; Tempst, P.; Snyder, S.H. Protein S-nitrosylation: A physiological signal for neuronal nitric oxide. Nat. Cell Biol. 2001, 3, 193–197.
- Anand, P.; Hausladen, A.; Wang, Y.J.; Zhang, G.F.; Stomberski, C.; Brunengraber, H.; Hess, D.T.; Stamler, J.S. Identification of S-nitroso-CoA reductases that regulate protein S-nitrosylation. Proc. Natl. Acad. Sci. USA 2014, 111, 18572–18577.
- Sircar, E.; Rai, S.R.; Wilson, M.A.; Schlossmacher, M.G.; Sengupta, R. Neurodegeneration: Impact of S-nitrosylated Parkin, DJ-1 and PINK1 on the pathogenesis of Parkinson’s disease. Arch. Biochem. Biophys. 2021, 704, 108869.
- Seth, D.; Hess, D.T.; Hausladen, A.; Wang, L.; Wang, Y.J.; Stamler, J.S. A Multiplex Enzymatic Machinery for Cellular Protein S-nitrosylation. Mol. Cell 2018, 69, 451–464.e6.
- Stomberski, C.T.; Hess, D.T.; Stamler, J.S. Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling. Antioxid. Redox Signal. 2019, 30, 1331–1351.
- Ballatori, N.; Krance, S.M.; Marchan, R.; Hammond, C.L. Plasma membrane glutathione transporters and their roles in cell physiology and pathophysiology. Mol. Aspects Med. 2009, 30, 13–28.
- Benhar, M.; Forrester, M.T.; Stamler, J.S. Protein denitrosylation: Enzymatic mechanisms and cellular functions. Nat. Rev. Mol. Cell Biol. 2009, 10, 721–732.
- Jensen, D.E.; Belka, G.K.; Du Bois, G.C. S-Nitrosoglutathione is a substrate for rat alcohol dehydrogenase class III isoenzyme. Biochem. J. 1998, 331, 659–668.
- Liu, L.; Hausladen, A.; Zeng, M.; Que, L.; Heitman, J.; Stamler, J.S. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature 2001, 410, 490–494.
- Bateman, R.L.; Rauh, D.; Tavshanjian, B.; Shokat, K.M. Human carbonyl reductase 1 is an S-nitrosoglutathione reductase. J. Biol. Chem. 2008, 283, 35756–35762.
- Rizza, S.; Di Leo, L.; Pecorari, C.; Giglio, P.; Faienza, F.; Montagna, C.; Maiani, E.; Puglia, M.; Bosisio, F.M.; Petersen, T.S.; et al. GSNOR deficiency promotes tumor growth via FAK1 S-nitrosylation. Cell Rep. 2023, 42, 111997.
- Nakamura, T.; Oh, C.K.; Zhang, X.; Lipton, S.A. Protein S-nitrosylation and oxidation contribute to protein misfolding in neurodegeneration. Free Radic. Biol. Med. 2021, 172, 562–577.
- Vrettou, S.; Wirth, B. S-Glutathionylation and S-Nitrosylation in Mitochondria: Focus on Homeostasis and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 15849.
- Jyotsana, N.; Ta, K.T.; DelGiorno, K.E. The Role of Cystine/Glutamate Antiporter SLC7A11/xCT in the Pathophysiology of Cancer. Front. Oncol. 2022, 12, 858462.
- Reichert, C.O.; de Freitas, F.A.; Sampaio-Silva, J.; Rokita-Rosa, L.; Barros, P.L.; Levy, D.; Bydlowski, S.P. Ferroptosis Mechanisms Involved in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8765.
- Palaniyappan, L.; Park, M.T.M.; Jeon, P.; Limongi, R.; Yang, K.; Sawa, A.; Théberge, J. Is There a Glutathione Centered Redox Dysregulation Subtype of Schizophrenia? Antioxidants 2021, 10, 1703.
- Peeples, E.S.; Genaro-Mattos, T.C. Ferroptosis: A Promising Therapeutic Target for Neonatal Hypoxic-Ischemic Brain Injury. Int. J. Mol. Sci. 2022, 23, 7420.
- Gleitze, S.; Paula-Lima, A.; Núñez, M.T.; Hidalgo, C. The calcium-iron connection in ferroptosis-mediated neuronal death. Free Radic. Biol. Med. 2021, 175, 28–41.
- Chen, K.; Jiang, X.; Wu, M.; Cao, X.; Bao, W.; Zhu, L.Q. Ferroptosis, a Potential Therapeutic Target in Alzheimer’s Disease. Front. Cell Dev. Biol. 2021, 9, 704298.
- Jakaria, M.; Belaidi, A.A.; Bush, A.I.; Ayton, S. Ferroptosis as a mechanism of neurodegeneration in Alzheimer’s disease. J. Neurochem. 2021, 159, 804–825.
- Crapper-McLachlan., D.R.; Dalton, A.J.; Kruck, T.P.; Bell, M.Y.; Smith, W.L.; Kalow, W.; Andrews, D.F. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 1991, 337, 1304–1308.
- Dysken, M.W.; Sano, M.; Asthana, S.; Vertrees, J.E.; Pallaki, M.; Llorente, M.; Love, S.; Schellenberg, G.D.; McCarten, J.R.; Malphurs, J.; et al. Effect of vitamin E and memantine on functional decline in Alzheimer disease: The TEAM-AD VA cooperative randomized trial. JAMA 2014, 311, 33–44.
- Da Costa, M.; Bernardi, J.; Costa, L.; Fiuza, T.; Brandão, R.; Ribeiro, M.F.; Amaral, J.D.; Rodrigues, C.M.P.; Pereira, M.E. N-acetylcysteine treatment attenuates the cognitive impairment and synaptic plasticity loss induced by streptozotocin. Chem. Biol. Interact. 2017, 272, 37–46.
- More, J.; Galusso, N.; Veloso, P.; Montecinos, L.; Finkelstein, J.P.; Sanchez, G.; Bull, R.; Valdés, J.L.; Hidalgo, C.; Paula-Lima, A. N-Acetylcysteine Prevents the Spatial Memory Deficits and the Redox-Dependent RyR2 Decrease Displayed by an Alzheimer’s Disease Rat Model. Front. Aging Neurosci. 2018, 10, 399.
- Stapper, Z.A.; Jahn, T.R. Changes in Glutathione Redox Potential Are Linked to Aβ42-Induced Neurotoxicity. Cell Rep. 2018, 24, 1696–1703.
- Guiney, S.J.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem. Int. 2017, 104, 34–48.
- Lin, K.J.; Chen, S.D.; Lin, K.L.; Liou, C.W.; Lan, M.Y.; Chuang, Y.C.; Wang, P.W.; Lee, J.J.; Wang, F.S.; Lin, H.Y.; et al. Iron Brain Menace: The Involvement of Ferroptosis in Parkinson Disease. Cells 2022, 11, 3829.
- Thapa, K.; Khan, H.; Kanojia, N.; Singh, T.G.; Kaur, A.; Kaur, G. Therapeutic Insights on Ferroptosis in Parkinson’s disease. Eur. J. Pharmacol. 2022, 930, 175133.
- Bisaglia, M.; Soriano, M.E.; Arduini, I.; Mammi, S.; Bubacco, L. Molecular characterization of dopamine-derived quinones reactivity toward NADH and glutathione: Implications for mitochondrial dysfunction in Parkinson disease. Biochim. Biophys. Acta 2010, 1802, 699–706.
- Segura-Aguilar, J.; Paris, I.; Muñoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915.
- Asanuma, M.; Miyazaki, I. Glutathione and Related Molecules in Parkinsonism. Int. J. Mol. Sci. 2021, 22, 8689.
- Cole, S.P.; Deeley, R.G. Transport of glutathione and glutathione conjugates by MRP1. Trends Pharmacol. Sci. 2006, 27, 438–446.
- Badillo-Ramírez, I.; Saniger, J.M.; Rivas-Arancibia, S. 5-S-cysteinyl-dopamine, a neurotoxic endogenous metabolite of dopamine: Implications for Parkinson’s disease. Neurochem. Int. 2019, 129, 104514.
- Stern, S.T.; Bruno, M.K.; Hennig, G.E.; Horton, R.A.; Roberts, J.C.; Cohen, S.D. Contribution of acetaminophen-cysteine to acetaminophen nephrotoxicity in CD-1 mice: I. Enhancement of acetaminophen nephrotoxicity by acetaminophen-cysteine. Toxicol. Appl. Pharmacol. 2005, 202, 151–159.
- Kim, K. Glutathione in the Nervous System as a Potential Therapeutic Target to Control the Development and Progression of Amyotrophic Lateral Sclerosis. Antioxidants 2021, 10, 1011.
- Valle, C.; Carrì, M.T. Cysteine Modifications in the Pathogenesis of ALS. Front. Mol. Neurosci. 2017, 10, 5.
- Pang, Y.; Liu, X.; Wang, X.; Shi, X.; Ma, L.; Zhang, Y.; Zhou, T.; Zhao, C.; Zhang, X.; Fan, B.; et al. Edaravone Modulates Neuronal GPX4/ACSL4/5-LOX to Promote Recovery After Spinal Cord Injury. Front. Cell Dev. Biol. 2022, 10, 849854.
- Watanabe, K.; Tanaka, M.; Yuki, S.; Hirai, M.; Yamamoto, Y. How is edaravone effective against acute ischemic stroke and amyotrophic lateral sclerosis? J. Clin. Biochem. Nutr. 2018, 62, 20–38.
- Spasić, S.; Nikolić-Kokić, A.; Miletić, S.; Oreščanin-Dušić, Z.; Spasić, M.B.; Blagojević, D.; Stević, Z. Edaravone May Prevent Ferroptosis in ALS. Curr. Drug Targets 2020, 21, 776–780.
- Stipanuk, M.H.; Ueki, I.; Dominy, J.E., Jr.; Simmons, C.R.; Hirschberger, L.L. Cysteine dioxygenase: A robust system for regulation of cellular cysteine levels. Amino Acids 2009, 37, 55–63.
- Wright, C.E.; Tallan, H.H.; Lin, Y.Y.; Gaull, G.E. Taurine: Biological update. Annu. Rev. Biochem. 1986, 55, 427–453.
- Chen, Q.; Konrad, C.; Sandhu, D.; Roychoudhury, D.; Schwartz, B.I.; Cheng, R.R.; Bredvik, K.; Kawamata, H.; Calder, E.L.; Studer, L.; et al. Accelerated transsulfuration metabolically defines a discrete subclass of amyotrophic lateral sclerosis patients. Neurobiol. Dis. 2020, 144, 105025.
- Varga, V.; Török, K.; Feuer, L.; Gulyás, J.; Somogyi, J. γ-Glutamyltransferase in the brain and its role in formation of γ-L-glutamyl-taurine. Prog. Clin. Biol. Res. 1985, 179, 115–125.
- Kobayashi, S.; Tokairin, Y.; Miyakoshi, T.; Saito, T.; Nagaoka, T.; Ikeda, Y.; Fujii, J.; Konno, H. Quantitative analysis of γ-glutamylpeptides by liquid chromatography-mass spectrometry and application for γ-glutamyltransferase assays. Anal. Biochem. 2019, 578, 13–22.
- Kobayashi, S.; Ikeda, Y.; Shigeno, Y.; Konno, H.; Fujii, J. γ-Glutamylcysteine synthetase and γ-glutamyl transferase as differential enzymatic sources of γ-glutamylpeptides in mice. Amino Acids 2020, 52, 555–566.
- Varga, V.; Janáky, R.; Marnela, K.M.; Saransaari, P.; Oja, S.S. Interactions of gamma-L-glutamyltaurine with excitatory aminoacidergic neurotransmission. Neurochem. Res. 1994, 19, 243–248.
- Uemura, S.; Ienaga, K.; Higashiura, K.; Kimura, H. Gamma-glutamyltaurine has potent and long-lasting antiepileptic action as demonstrated by intra-amygdaloid injection in amygdala-kindled rats. Brain Res. 1992, 594, 347–350.
- Bittner, S.; Win, T.; Gupta, R. γ-L-glutamyltaurine. Amino Acids 2005, 28, 343–356.
- Fujii, J.; Homma, T.; Kobayashi, S. Ferroptosis caused by cysteine insufficiency and oxidative insult. Free Radic. Res. 2020, 54, 969–980.
- Griffith, O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med. 1999, 27, 922–935.
- Orlowski, M.; Wilk, S. Intermediates of the gamma-glutamyl cycle in mouse tissues. Influence of administration of amino acids on pyrrolidone carboxylate and gamma-glutamyl amino acids. Eur. J. Biochem. 1975, 53, 581–590.
- Soga, T.; Baran, R.; Suematsu, M.; Ueno, Y.; Ikeda, S.; Sakurakawa, T.; Kakazu, Y.; Ishikawa, T.; Robert, M.; Nishioka, T.; et al. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J. Biol. Chem. 2006, 281, 16768–16776.
- Kobayashi, S.; Lee, J.; Takao, T.; Fujii, J. Increased ophthalmic acid production is supported by amino acid catabolism under fasting conditions in mice. Biochem. Biophys. Res. Commun. 2017, 491, 649–655.
- Soga, T.; Sugimoto, M.; Honma, M.; Mori, M.; Igarashi, K.; Kashikura, K.; Ikeda, S.; Hirayama, A.; Yamamoto, T.; Yoshida, H.; et al. Serum metabolomics reveals γ-glutamyl dipeptides as biomarkers for discrimination among different forms of liver disease. J. Hepatol. 2011, 55, 896–905.
- Deepmala; Slattery, J.; Kumar, N.; Delhey, L.; Berk, M.; Dean, O.; Spielholz, C.; Frye, R. Clinical trials of N-acetylcysteine in psychiatry and neurology: A systematic review. Neurosci. Biobehav. Rev. 2015, 55, 294–321.
- Kang, Y.P.; Mockabee-Macias, A.; Jiang, C.; Falzone, A.; Prieto-Farigua, N.; Stone, E.; Harris, I.S.; DeNicola, G.M. Non-canonical Glutamate-Cysteine Ligase Activity Protects against Ferroptosis. Cell Metab. 2021, 33, 174–189.e7.
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.
- Homma, T.; Kobayashi, S.; Fujii, J. Methionine Deprivation Reveals the Pivotal Roles of Cell Cycle Progression in Ferroptosis That Is Induced by Cysteine Starvation. Cells 2022, 11, 1603.
- Maclean, K.N.; Jiang, H.; Aivazidis, S.; Kim, E.; Shearn, C.T.; Harris, P.S.; Petersen, D.R.; Allen, R.H.; Stabler, S.P.; Roede, J.R. Taurine treatment prevents derangement of the hepatic γ-glutamyl cycle and methylglyoxal metabolism in a mouse model of classical homocystinuria: Regulatory crosstalk between thiol and sulfinic acid metabolism. FASEB J. 2018, 32, 1265–1280.
- Li, H.Q.; Xia, S.N.; Xu, S.Y.; Liu, P.Y.; Gu, Y.; Bao, X.Y.; Xu, Y.; Cao, X. γ-Glutamylcysteine Alleviates Ischemic Stroke-Induced Neuronal Apoptosis by Inhibiting ROS-Mediated Endoplasmic Reticulum Stress. Oxid. Med. Cell Longev. 2021, 2021, 2961079.
- Zhang, H.; Kovacs-Nolan, J.; Kodera, T.; Eto, Y.; Mine, Y. γ-Glutamyl cysteine and γ-glutamyl valine inhibit TNF-α signaling in intestinal epithelial cells and reduce inflammation in a mouse model of colitis via allosteric activation of the calcium-sensing receptor. Biochim. Biophys. Acta 2015, 1852, 792–804.
- Hewson, C.K.; Capraro, A.; Wong, S.L.; Pandzic, E.; Zhong, L.; Fernando, B.S.M.; Awatade, N.T.; Hart-Smith, G.; Whan, R.M.; Thomas, S.R.; et al. Novel Antioxidant Therapy with the Immediate Precursor to Glutathione, γ-Glutamylcysteine (GGC), Ameliorates LPS-Induced Cellular Stress in In Vitro 3D-Differentiated Airway Model from Primary Cystic Fibrosis Human Bronchial Cells. Antioxidants 2020, 9, 1204.
- Bi, A.; Wang, Y.; Chen, L.; Yin, Z.; Luo, L. γ-Glutamylcysteine attenuates amyloid-β oligomers-induced neuroinflammation in microglia via blocking NF-κB signaling pathway. Chem. Biol. Interact. 2022, 363, 110019.
- Zhou, J.; Yan, X.; Bi, X.; Lu, S.; Liu, X.; Yang, C.; Shi, Y.; Luo, L.; Yin, Z. γ-Glutamylcysteine rescues mice from TNBS-driven inflammatory bowel disease through regulating macrophages polarization. Inflamm. Res. 2023, 72, 603–621.
- Liu, Y.; Chen, Z.; Li, B.; Yao, H.; Zarka, M.; Welch, J.; Sachdev, P.; Bridge, W.; Braidy, N. Supplementation with γ-glutamylcysteine (γ-GC) lessens oxidative stress, brain inflammation and amyloid pathology and improves spatial memory in a murine model of AD. Neurochem. Int. 2021, 144, 104931.
- Zhang, R.; Lei, J.; Chen, L.; Wang, Y.; Yang, G.; Yin, Z.; Luo, L. γ-Glutamylcysteine Exerts Neuroprotection Effects against Cerebral Ischemia/Reperfusion Injury through Inhibiting Lipid Peroxidation and Ferroptosis. Antioxidants 2022, 11, 1653.
- Ferguson, G.; Bridge, W. Glutamate cysteine ligase and the age-related decline in cellular glutathione: The therapeutic potential of γ-glutamylcysteine. Arch. Biochem. Biophys. 2016, 593, 12–23.
- Zarka, M.H.; Bridge, W.J. Oral administration of γ-glutamylcysteine increases intracellular glutathione levels above homeostasis in a randomised human trial pilot study. Redox Biol. 2017, 11, 631–636.
- Yang, Y.; Li, L.; Hang, Q.; Fang, Y.; Dong, X.; Cao, P.; Yin, Z.; Luo, L. γ-glutamylcysteine exhibits anti-inflammatory effects by increasing cellular glutathione level. Redox Biol. 2019, 20, 157–166.
- Quintana-Cabrera, R.; Fernandez-Fernandez, S.; Bobo-Jimenez, V.; Escobar, J.; Sastre, J.; Almeida, A.; Bolaños, J.P. γ-Glutamylcysteine detoxifies reactive oxygen species by acting as glutathione peroxidase-1 cofactor. Nat. Commun. 2012, 3, 718.
- Wang, M.; Yao, Y.; Kuang, D.; Hampson, D.R. Activation of family C G-protein-coupled receptors by the tripeptide glutathione. J. Biol. Chem. 2006, 281, 8864–8870.
- Goralski, T.; Ram, J.L. Extracellular Calcium Receptor as a Target for Glutathione and Its Derivatives. Int. J. Mol. Sci. 2022, 23, 717.
- Bandyopadhyay, S.; Tfelt-Hansen, J.; Chattopadhyay, N. Diverse roles of extracellular calcium-sensing receptor in the central nervous system. J. Neurosci Res. 2010, 88, 2073–2082.
- Brennan, S.C.; Davies, T.S.; Schepelmann, M.; Riccardi, D. Emerging roles of the extracellular calcium-sensing receptor in nutrient sensing: Control of taste modulation and intestinal hormone secretion. Br. J. Nutr. 2014, 111, S16–S22.
- Guha, S.; Majumder, K. J Comprehensive Review of γ-Glutamyl Peptides (γ-GPs) and Their Effect on Inflammation Concerning Cardiovascular Health. Agric. Food Chem. 2022, 70, 7851–7870.
- Giudice, M.L.; Mihalik, B.; Dinnyés, A.; Kobolák, J. The Nervous System Relevance of the Calcium Sensing Receptor in Health and Disease. Molecules 2019, 24, 2546.
- Iamartino, L.; Brandi, M.L. The calcium-sensing receptor in inflammation: Recent updates. Front. Physiol. 2022, 13, 1059369.
- Chiarini, A.; Armato, U.; Gardenal, E.; Gui, L.; Dal Prà, I. Amyloid β-Exposed Human Astrocytes Overproduce Phospho-Tau and Overrelease It within Exosomes, Effects Suppressed by Calcilytic NPS 2143-Further Implications for Alzheimer’s Therapy. Front. Neurosci. 2017, 11, 217.
- Dal Prà, I.; Armato, U.; Chiarini, A. Family C G-Protein-Coupled Receptors in Alzheimer’s Disease and Therapeutic Implications. Front. Pharmacol. 2019, 10, 1282.