 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Juan José Garrido | -- | 2804 | 2023-05-05 12:39:11 | | | |
| 2 | Dean Liu | Meta information modification | 2804 | 2023-05-06 07:10:39 | | |
Video Upload Options
Brain channelopathies are a group of neurological disorders that result from genetic mutations affecting ion channels in the brain. Ion channels are specialized proteins that play a crucial role in the electrical activity of nerve cells by controlling the flow of ions such as sodium, potassium, and calcium. When these channels are not functioning properly, they can cause a wide range of neurological symptoms such as seizures, movement disorders, and cognitive impairment. In this context, the axon initial segment (AIS) is the site of action potential initiation in most neurons. This region is characterized by a high density of voltage-gated sodium channels (VGSCs), which are responsible for the rapid depolarization that occurs when the neuron is stimulated. The AIS is also enriched in other ion channels, such as potassium channels, that play a role in shaping the action potential waveform and determining the firing frequency of the neuron. In addition to ion channels, the AIS contains a complex cytoskeletal structure that helps to anchor the channels in place and regulate their function.
1. Introduction
1.1. Axon Initial Segment Structure and Physiology
1.2. Axon Initial Segment Composition and Structure
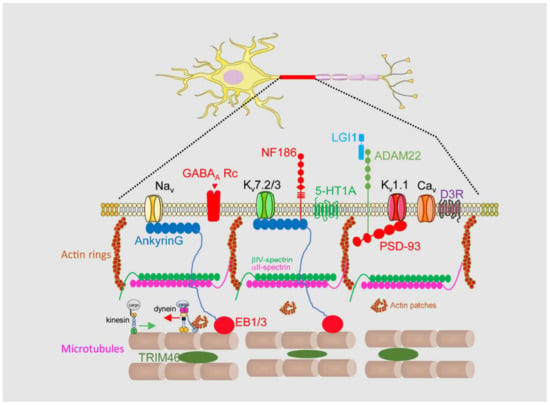
2. Axon Initial Segment, Mental Disorders, and Neurodegenerative Diseases
2.1. Mental Disorders
2.2. Neurodevelopmental Disorders
2.3. Neuroinflammatory Diseases and Glial Cells
2.4. Autoimmune Diseases
2.5. Alzheimer’s Disease
References
- Bender, K.J.; Trussell, L.O. The physiology of the axon initial segment. Annu. Rev. Neurosci. 2012, 35, 249–265.
- Kole, M.H.; Ilschner, S.U.; Kampa, B.M.; Williams, S.R.; Ruben, P.C.; Stuart, G.J. Action potential generation requires a high sodium channel density in the axon initial segment. Nat. Neurosci. 2008, 11, 178–186.
- Del Puerto, A.; Fronzaroli-Molinieres, L.; Perez-Alvarez, M.J.; Giraud, P.; Carlier, E.; Wandosell, F.; Debanne, D.; Garrido, J.J. ATP-P2X7 Receptor Modulates Axon Initial Segment Composition and Function in Physiological Conditions and Brain Injury. Cereb. Cortex 2015, 25, 2282–2294.
- Grubb, M.S.; Burrone, J. Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature 2010, 465, 1070–1074.
- Kuba, H. Plasticity at the axon initial segment. Commun. Integr. Biol. 2010, 3, 597–598.
- Rasband, M.N. The axon initial segment and the maintenance of neuronal polarity. Nat. Rev. Neurosci. 2010, 11, 552–562.
- Schafer, D.P.; Jha, S.; Liu, F.; Akella, T.; McCullough, L.D.; Rasband, M.N. Disruption of the axon initial segment cytoskeleton is a new mechanism for neuronal injury. J. Neurosci. 2009, 29, 13242–13254.
- Jenkins, S.M.; Bennett, V. Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J. Cell Biol. 2001, 155, 739–746.
- Ogawa, Y.; Horresh, I.; Trimmer, J.S.; Bredt, D.S.; Peles, E.; Rasband, M.N. Postsynaptic density-93 clusters Kv1 channels at axon initial segments independently of Caspr2. J. Neurosci. 2008, 28, 5731–5739.
- Nakada, C.; Ritchie, K.; Oba, Y.; Nakamura, M.; Hotta, Y.; Iino, R.; Kasai, R.S.; Yamaguchi, K.; Fujiwara, T.; Kusumi, A. Accumulation of anchored proteins forms membrane diffusion barriers during neuronal polarization. Nat. Cell Biol. 2003, 5, 626–632.
- Leterrier, C.; Vacher, H.; Fache, M.P.; d’Ortoli, S.A.; Castets, F.; Autillo-Touati, A.; Dargent, B. End-binding proteins EB3 and EB1 link microtubules to ankyrin G in the axon initial segment. Proc. Natl. Acad. Sci. USA 2011, 108, 8826–8831.
- Leterrier, C. The Axon Initial Segment: An Updated Viewpoint. J. Neurosci. 2018, 38, 2135–2145.
- Sanchez-Ponce, D.; Munoz, A.; Garrido, J.J. Casein kinase 2 and microtubules control axon initial segment formation. Mol. Cell. Neurosci. 2011, 46, 222–234.
- Konishi, Y.; Setou, M. Tubulin tyrosination navigates the kinesin-1 motor domain to axons. Nat. Neurosci. 2009, 12, 559–567.
- Tapia, M.; Wandosell, F.; Garrido, J.J. Impaired function of HDAC6 slows down axonal growth and interferes with axon initial segment development. PLoS ONE 2010, 5, e12908.
- Harterink, M.; Vocking, K.; Pan, X.; Soriano Jerez, E.M.; Slenders, L.; Freal, A.; Tas, R.P.; van de Wetering, W.J.; Timmer, K.; Motshagen, J.; et al. TRIM46 Organizes Microtubule Fasciculation in the Axon Initial Segment. J. Neurosci. 2019, 39, 4864–4873.
- Papandreou, M.J.; Leterrier, C. The functional architecture of axonal actin. Mol. Cell. Neurosci. 2018, 91, 151–159.
- Eichel, K.; Shen, K. The function of the axon initial segment in neuronal polarity. Dev. Biol. 2022, 489, 47–54.
- Kuijpers, M.; van de Willige, D.; Freal, A.; Chazeau, A.; Franker, M.A.; Hofenk, J.; Rodrigues, R.J.; Kapitein, L.C.; Akhmanova, A.; Jaarsma, D.; et al. Dynein Regulator NDEL1 Controls Polarized Cargo Transport at the Axon Initial Segment. Neuron 2016, 89, 461–471.
- Benedeczky, I.; Molnar, E.; Somogyi, P. The cisternal organelle as a Ca(2+)-storing compartment associated with GABAergic synapses in the axon initial segment of hippocampal pyramidal neurones. Exp. Brain Res. 1994, 101, 216–230.
- Schluter, A.; Del Turco, D.; Deller, T.; Gutzmann, A.; Schultz, C.; Engelhardt, M. Structural Plasticity of Synaptopodin in the Axon Initial Segment during Visual Cortex Development. Cereb. Cortex 2017, 27, 4662–4675.
- Hedstrom, K.L.; Ogawa, Y.; Rasband, M.N. AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J. Cell Biol. 2008, 183, 635–640.
- Sobotzik, J.M.; Sie, J.M.; Politi, C.; Del Turco, D.; Bennett, V.; Deller, T.; Schultz, C. AnkyrinG is required to maintain axo-dendritic polarity in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 17564–17569.
- Garrido, J.J.; Giraud, P.; Carlier, E.; Fernandes, F.; Moussif, A.; Fache, M.P.; Debanne, D.; Dargent, B. A targeting motif involved in sodium channel clustering at the axonal initial segment. Science 2003, 300, 2091–2094.
- Pan, Z.; Kao, T.; Horvath, Z.; Lemos, J.; Sul, J.Y.; Cranstoun, S.D.; Bennett, V.; Scherer, S.S.; Cooper, E.C. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J. Neurosci. 2006, 26, 2599–2613.
- Zhou, D.; Lambert, S.; Malen, P.L.; Carpenter, S.; Boland, L.M.; Bennett, V. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J. Cell Biol. 1998, 143, 1295–1304.
- Smolin, B.; Karry, R.; Gal-Ben-Ari, S.; Ben-Shachar, D. Differential expression of genes encoding neuronal ion-channel subunits in major depression, bipolar disorder and schizophrenia: Implications for pathophysiology. Int. J. Neuropsychopharmacol. 2012, 15, 869–882.
- Imbrici, P.; Camerino, D.C.; Tricarico, D. Major channels involved in neuropsychiatric disorders and therapeutic perspectives. Front. Genet. 2013, 4, 76.
- Bavamian, S.; Mellios, N.; Lalonde, J.; Fass, D.M.; Wang, J.; Sheridan, S.D.; Madison, J.M.; Zhou, F.; Rueckert, E.H.; Barker, D.; et al. Dysregulation of miR-34a links neuronal development to genetic risk factors for bipolar disorder. Mol. Psychiatry 2015, 20, 573–584.
- Yang, R.; Walder-Christensen, K.K.; Lalani, S.; Yan, H.; Garcia-Prieto, I.D.; Alvarez, S.; Fernandez-Jaen, A.; Speltz, L.; Jiang, Y.H.; Bennett, V. Neurodevelopmental mutation of giant ankyrin-G disrupts a core mechanism for axon initial segment assembly. Proc. Natl. Acad. Sci. USA 2019, 116, 19717–19726.
- Zhu, S.; Cordner, Z.A.; Xiong, J.; Chiu, C.T.; Artola, A.; Zuo, Y.; Nelson, A.D.; Kim, T.Y.; Zaika, N.; Woolums, B.M.; et al. Genetic disruption of ankyrin-G in adult mouse forebrain causes cortical synapse alteration and behavior reminiscent of bipolar disorder. Proc. Natl. Acad. Sci. USA 2017, 114, 10479–10484.
- Rees, E.; Carrera, N.; Morgan, J.; Hambridge, K.; Escott-Price, V.; Pocklington, A.J.; Richards, A.L.; Pardinas, A.F.; Investigators, G.; McDonald, C.; et al. Targeted Sequencing of 10,198 Samples Confirms Abnormalities in Neuronal Activity and Implicates Voltage-Gated Sodium Channels in Schizophrenia Pathogenesis. Biol. Psychiatry 2019, 85, 554–562.
- Mi, Z.; Yang, J.; He, Q.; Zhang, X.; Xiao, Y.; Shu, Y. Alterations of Electrophysiological Properties and Ion Channel Expression in Prefrontal Cortex of a Mouse Model of Schizophrenia. Front. Cell. Neurosci. 2019, 13, 554.
- Page, S.C.; Sripathy, S.R.; Farinelli, F.; Ye, Z.; Wang, Y.; Hiler, D.J.; Pattie, E.A.; Nguyen, C.V.; Tippani, M.; Moses, R.L.; et al. Electrophysiological measures from human iPSC-derived neurons are associated with schizophrenia clinical status and predict individual cognitive performance. Proc. Natl. Acad. Sci. USA 2022, 119, e2109395119.
- Lam, M.; Chen, C.Y.; Li, Z.; Martin, A.R.; Bryois, J.; Ma, X.; Gaspar, H.; Ikeda, M.; Benyamin, B.; Brown, B.C.; et al. Comparative genetic architectures of schizophrenia in East Asian and European populations. Nat. Genet. 2019, 51, 1670–1678.
- Kaphzan, H.; Buffington, S.A.; Jung, J.I.; Rasband, M.N.; Klann, E. Alterations in intrinsic membrane properties and the axon initial segment in a mouse model of Angelman syndrome. J. Neurosci. 2011, 31, 17637–17648.
- Booker, S.A.; Simoes de Oliveira, L.; Anstey, N.J.; Kozic, Z.; Dando, O.R.; Jackson, A.D.; Baxter, P.S.; Isom, L.L.; Sherman, D.L.; Hardingham, G.E.; et al. Input-Output Relationship of CA1 Pyramidal Neurons Reveals Intact Homeostatic Mechanisms in a Mouse Model of Fragile X Syndrome. Cell Rep. 2020, 32, 107988.
- Tian, T.; Quintana-Urzainqui, I.; Kozic, Z.; Pratt, T.; Price, D.J. Pax6 loss alters the morphological and electrophysiological development of mouse prethalamic neurons. Development 2022, 149, dev200052.
- Jacko, M.; Weyn-Vanhentenryck, S.M.; Smerdon, J.W.; Yan, R.; Feng, H.; Williams, D.J.; Pai, J.; Xu, K.; Wichterle, H.; Zhang, C. Rbfox Splicing Factors Promote Neuronal Maturation and Axon Initial Segment Assembly. Neuron 2018, 97, 853–868.e6.
- Hong, T.; Falcone, C.; Dufour, B.; Amina, S.; Castro, R.P.; Regalado, J.; Pearson, W.; Noctor, S.C.; Martinez-Cerdeno, V. GABA(A)Ralpha2 is Decreased in the Axon Initial Segment of Pyramidal Cells in Specific Areas of the Prefrontal Cortex in Autism. Neuroscience 2020, 437, 76–86.
- Usui, N.; Tian, X.; Harigai, W.; Togawa, S.; Utsunomiya, R.; Doi, T.; Miyoshi, K.; Shinoda, K.; Tanaka, J.; Shimada, S.; et al. Length impairments of the axon initial segment in rodent models of attention-deficit hyperactivity disorder and autism spectrum disorder. Neurochem. Int. 2022, 153, 105273.
- Baalman, K.L.; Cotton, R.J.; Rasband, S.N.; Rasband, M.N. Blast wave exposure impairs memory and decreases axon initial segment length. J. Neurotrauma 2013, 30, 741–751.
- Craner, M.J.; Newcombe, J.; Black, J.A.; Hartle, C.; Cuzner, M.L.; Waxman, S.G. Molecular changes in neurons in multiple sclerosis: Altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc. Natl. Acad. Sci. USA 2004, 101, 8168–8173.
- Jorgensen, H.S.; Jensen, D.B.; Dimintiyanova, K.P.; Bonnevie, V.S.; Hedegaard, A.; Lehnhoff, J.; Moldovan, M.; Grondahl, L.; Meehan, C.F. Increased Axon Initial Segment Length Results in Increased Na(+) Currents in Spinal Motoneurones at Symptom Onset in the G127X SOD1 Mouse Model of Amyotrophic Lateral Sclerosis. Neuroscience 2021, 468, 247–264.
- Sasaki, S.; Warita, H.; Abe, K.; Iwata, M. Impairment of axonal transport in the axon hillock and the initial segment of anterior horn neurons in transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol. 2005, 110, 48–56.
- Senol, A.D.; Pinto, G.; Beau, M.; Guillemot, V.; Dupree, J.L.; Stadelmann, C.; Ranft, J.; Lubetzki, C.; Davenne, M. Alterations of the axon initial segment in multiple sclerosis grey matter. Brain Commun. 2022, 4, fcac284.
- Baalman, K.; Marin, M.A.; Ho, T.S.; Godoy, M.; Cherian, L.; Robertson, C.; Rasband, M.N. Axon initial segment-associated microglia. J. Neurosci. 2015, 35, 2283–2292.
- Gallo, N.B.; Berisha, A.; Van Aelst, L. Microglia regulate chandelier cell axo-axonic synaptogenesis. Proc. Natl. Acad. Sci. USA 2022, 119, e2114476119.
- Schmaul, S.; Hanuscheck, N.; Bittner, S. Astrocytic potassium and calcium channels as integrators of the inflammatory and ischemic CNS microenvironment. Biol. Chem. 2021, 402, 1519–1530.
- Lezmy, J.; Arancibia-Carcamo, I.L.; Quintela-Lopez, T.; Sherman, D.L.; Brophy, P.J.; Attwell, D. Astrocyte Ca(2+)-evoked ATP release regulates myelinated axon excitability and conduction speed. Science 2021, 374, eabh2858.
- Bartley, C.M.; Ngo, T.T.; Alvarenga, B.D.; Kung, A.F.; Teliska, L.H.; Sy, M.; DeRisi, J.L.; Rasband, M.N.; Pittock, S.J.; Dubey, D.; et al. betaIV-Spectrin Autoantibodies in 2 Individuals With Neuropathy of Possible Paraneoplastic Origin: A Case Series. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e1188.
- Berghs, S.; Ferracci, F.; Maksimova, E.; Gleason, S.; Leszczynski, N.; Butler, M.; De Camilli, P.; Solimena, M. Autoimmunity to beta IV spectrin in paraneoplastic lower motor neuron syndrome. Proc. Natl. Acad. Sci. USA 2001, 98, 6945–6950.
- Valencia-Sanchez, C.; Knight, A.M.; Hammami, M.B.; Guo, Y.; Mills, J.R.; Kryzer, T.J.; Piquet, A.L.; Amin, A.; Heinzelmann, M.; Lucchinetti, C.F.; et al. Characterisation of TRIM46 autoantibody-associated paraneoplastic neurological syndrome. J. Neurol. Neurosurg. Psychiatry 2022, 93, 196–200.
- Bartley, C.M.; Ngo, T.T.; Cadwell, C.R.; Harroud, A.; Schubert, R.D.; Alvarenga, B.D.; Hawes, I.A.; Zorn, K.C.; Hunyh, T.; Teliska, L.H.; et al. Dual ankyrinG and subpial autoantibodies in a man with well-controlled HIV infection with steroid-responsive meningoencephalitis: A case report. Front. Neurol. 2022, 13, 1102484.
- Ohkawa, T.; Fukata, Y.; Yamasaki, M.; Miyazaki, T.; Yokoi, N.; Takashima, H.; Watanabe, M.; Watanabe, O.; Fukata, M. Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J. Neurosci. 2013, 33, 18161–18174.
- Seagar, M.; Russier, M.; Caillard, O.; Maulet, Y.; Fronzaroli-Molinieres, L.; De San Feliciano, M.; Boumedine-Guignon, N.; Rodriguez, L.; Zbili, M.; Usseglio, F.; et al. LGI1 tunes intrinsic excitability by regulating the density of axonal Kv1 channels. Proc. Natl. Acad. Sci. USA 2017, 114, 7719–7724.
- Pinatel, D.; Hivert, B.; Boucraut, J.; Saint-Martin, M.; Rogemond, V.; Zoupi, L.; Karagogeos, D.; Honnorat, J.; Faivre-Sarrailh, C. Inhibitory axons are targeted in hippocampal cell culture by anti-Caspr2 autoantibodies associated with limbic encephalitis. Front. Cell. Neurosci. 2015, 9, 265.
- Zandi, M.S.; Irani, S.R.; Lang, B.; Waters, P.; Jones, P.B.; McKenna, P.; Coles, A.J.; Vincent, A.; Lennox, B.R. Disease-relevant autoantibodies in first episode schizophrenia. J. Neurol. 2011, 258, 686–688.
- Sun, X.; Wu, Y.; Gu, M.; Liu, Z.; Ma, Y.; Li, J.; Zhang, Y. Selective filtering defect at the axon initial segment in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. USA 2014, 111, 14271–14276.
- Vitale, P.; Salgueiro-Pereira, A.R.; Lupascu, C.A.; Willem, M.; Migliore, R.; Migliore, M.; Marie, H. Analysis of Age-Dependent Alterations in Excitability Properties of CA1 Pyramidal Neurons in an APPPS1 Model of Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 668948.
- Liu, C.; Tan, F.C.; Xiao, Z.C.; Dawe, G.S. Amyloid precursor protein enhances Nav1.6 sodium channel cell surface expression. J. Biol. Chem. 2015, 290, 12048–12057.
- Yuan, D.J.; Yang, G.; Wu, W.; Li, Q.F.; Xu, D.E.; Ntim, M.; Jiang, C.Y.; Liu, J.C.; Zhang, Y.; Wang, Y.Z.; et al. Reducing Nav1.6 expression attenuates the pathogenesis of Alzheimer’s disease by suppressing BACE1 transcription. Aging Cell 2022, 21, e13593.
- Hessler, S.; Zheng, F.; Hartmann, S.; Rittger, A.; Lehnert, S.; Volkel, M.; Nissen, M.; Edelmann, E.; Saftig, P.; Schwake, M.; et al. beta-Secretase BACE1 regulates hippocampal and reconstituted M-currents in a beta-subunit-like fashion. J. Neurosci. 2015, 35, 3298–3311.
- Di Benedetto, G.; Burgaletto, C.; Bellanca, C.M.; Munafo, A.; Bernardini, R.; Cantarella, G. Role of Microglia and Astrocytes in Alzheimer’s Disease: From Neuroinflammation to Ca(2+) Homeostasis Dysregulation. Cells 2022, 11, 2728.
- Lipkin, A.M.; Cunniff, M.M.; Spratt, P.W.E.; Lemke, S.M.; Bender, K.J. Functional Microstructure of Ca(V)-Mediated Calcium Signaling in the Axon Initial Segment. J. Neurosci. 2021, 41, 3764–3776.
- Anton-Fernandez, A.; Leon-Espinosa, G.; DeFelipe, J.; Munoz, A. Pyramidal cell axon initial segment in Alzheimer s disease. Sci. Rep. 2022, 12, 8722.
- Ma, F.; Akolkar, H.; Xu, J.; Liu, Y.; Popova, D.; Xie, J.; Youssef, M.M.; Benosman, R.; Hart, R.P.; Herrup, K. The Amyloid Precursor Protein Modulates the Position and Length of the Axon Initial Segment. J. Neurosci. 2023, 43, 1830–1844.
- Marin, M.A.; Ziburkus, J.; Jankowsky, J.; Rasband, M.N. Amyloid-beta plaques disrupt axon initial segments. Exp. Neurol. 2016, 281, 93–98.
- Sohn, P.D.; Huang, C.T.; Yan, R.; Fan, L.; Tracy, T.E.; Camargo, C.M.; Montgomery, K.M.; Arhar, T.; Mok, S.A.; Freilich, R.; et al. Pathogenic Tau Impairs Axon Initial Segment Plasticity and Excitability Homeostasis. Neuron 2019, 104, 458–470.e5.

