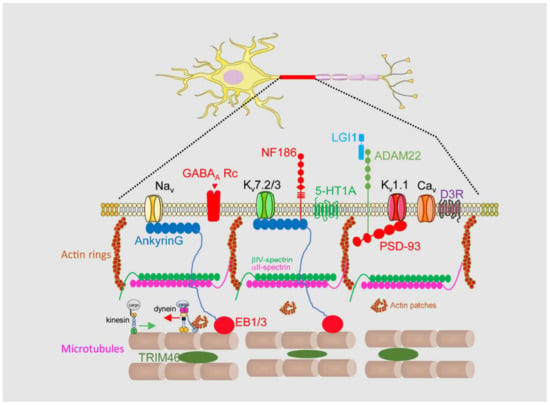
An AIS is a highly stable structure that is the first 20–60 μm of the axon (
Figure 1). Its function as a membrane diffusion barrier is due to a high concentration of membrane proteins such as neurofascin, NrCAM, TAG1, ADAM22, and voltage-gated ion channels [
15]. These membrane proteins are concentrated and tethered at the AIS through specific aminoacidic motifs that bind to AIS scaffold proteins such as ankyrinG, βIV-spectrin, or PSD-93 [
17,
18]. AIS proteins are organized into a complex network, which forms a diffusion barrier between the axonal and somatodendritic compartments of the neuron, thereby preventing the diffusion of proteins and lipids between these compartments [
19]. Among them, ankyrinG and βIV-spectrin are essential in maintaining AIS structure and integrity, linking membrane proteins to the AIS cytoskeleton. AnkyrinG interacts with microtubules through EB proteins [
20], while βIV-spectrin links ankyrinG to the actin cytoskeleton [
21]. Both actin and microtubules at the AIS contain differential characteristics compared to the somatodendritic or axonal cytoskeleton. Microtubules at the AIS contain more detyrosinated and acetylated tubulin [
22] and are more stable and act as a platform for axonal motor proteins [
23] such as kinesin-1. Post-translational modifications (PTMs) of tubulin, such as acetylation, alter the ankyrinG distribution and impair kinesin-1 entry to the axon [
6]. Tubulin PTMs confer a high degree of stability to the AIS, but additionally TRIM-46 (Tripartite motif containing 46) contributes to microtubule fasciculation [
24]. Different actin structures have been identified at the AIS, forming actin rings and actin patches. Actin rings are supported by αII- and βIV-spectrin tetramers and are thought to participate in the initial axon structural plasticity [
25], while actin patches may serve as a mechanism controlling the axonal entry of somatodendritic cargoes and their retrograde transport by dynein [
26,
27]. The AIS also contains an actin-related organelle, the cisternal organelle (CO), which is subject to structural changes associated with AIS plasticity during development and environmental changes [
28,
29]. Its function is unknown, but it was suggested to be putative Ca
2+ store involved in calcium regulation at the AIS.
The complexity and specificity of the AIS structure, together with the still limited information available about its protein composition, renders it difficult to completely understand how action potentials are regulated and how voltage-gated ion channel density and location contribute to this regulation.
2. Axon Initial Segment, Mental Disorders, and Neurodegenerative Diseases
2.1. Mental Disorders
Bipolar disorder is a mental health condition characterized by episodes of manic and depressive symptoms. The exact causes of bipolar disorder are not fully understood, but it is believed to involve a complex interplay of genetic, environmental, and neurobiological factors. Research has suggested that abnormalities in the function and structure of neurons, including those in the AIS, may contribute to the development of bipolar disorder. Gene array studies in bipolar disease patients have shown decreased expression of potassium channel K
v1.1, while K
v7.2 and K
v7.3 channels had increased expression [
98]. Different studies have highlighted the role of
ANK3 gene single nucleotide polymorphims (SNPs) as a risk factor for bipolar disorder. These SNPs affect ankyrinG expression and concomitantly the proper anchoring of potassium channels [
99]. Moreover,
ANK3 mRNA can be regulated by the expression level of microRNAs in bipolar disorder patients, such as miR34a and miR10b-5p [
100]. ankyrinG (480 kDa) is essential for AIS assembly and three missense mutations have been identified in humans. These mutations increase the AIS length, decrease the ankyrinG density, and prevent βIV spectrin recruitment. As a consequence, voltage-gated sodium channels are diffused along the longer AIS and action potential firing has a reduced temporal precision [
101]. Furthermore, fore-brain specific knockout of ankyrinG in adult mice shows characteristics reminiscent of aspects of human mania [
102].
Schizophrenia is a group of complex mental disorders characterized by psychotic, negative, and cognitive symptoms. Among other alterations, some studies have shown that individuals with schizophrenia have alterations in the expression and distribution of sodium channels in various regions of the brain, including the prefrontal cortex and hippocampus [
103]. Loss of function (LoF) mutations and rare coding variants (RCVs) in sodium channels increase the risk of schizophrenia [
103]. Mouse models related to schizophrenia have shown increased Na
v1.2 and Na
v1.6 channel expressions and changes in the electrophysiological properties of action potentials [
104,
105]. There is no clear evidence of the role of other potassium or calcium channels expressed in the AIS in schizophrenia. The link between
ANK3 and schizophrenia remains unclear. Some GWAS studies have proposed an association between
ANK3 SNPs and schizophrenia; however, a large GWAS did not show a significant association [
106].
2.2. Neurodevelopmental Disorders
Neuronal polarization, functional polarity, and brain network physiology depend on proper early AIS development and maturation. Several neurodevelopmental disorders have been related to early alterations in AIS development. The mouse model of Angelman syndrome exhibited a significant increase in the AIS length in hippocampal neurons [
88]. This syndrome is due to the loss of function of the
UBE3A gene and is associated with intellectual disability, epilepsy, ataxia, and autism. An increased length is accompanied by a higher expression of voltage-gated sodium channels and ankyrinG. The same length increase has been detected in a fragile X mouse model (Fmr1
−/y) that shows increased neuronal excitability [
107]. More recently, the loss of a transcription factor, Pax6, was found to change the length and position of the AIS in prethalamic neurons [
108]. Similarly, the loss of function of Rbfox proteins that regulate alternative splicing and posttranscriptional regulation leads to defects in AnkG localization and an immature electrophysiology [
109], showing the importance of AnkG developmental splicing. In fact, neurons that express ankyrinG, including a 33 nucleotide cassette upstream of the first ZU5 domain, do generate significantly less action potentials, indicating a lower density of voltage-gated ion channels [
101].
Autism spectrum disorders are also related to AIS alterations. Subjects with autism show a decreased GABA synthesis in the prefrontal cortex that can lead to downregulation of the GABAARa2 protein in the AIS of pyramidal neurons and affect the inhibitory control of action potentials, contributing to an excitation/inhibition imbalance [
110]. Autism mouse models also show a shortening of the AIS in the primary somatosensory barrel cortex, similar to the AIS length change in the same region in ADHD (attention-deficit hyperactivity disorder) model rats [
111]. The contribution of voltage-gated ion channels or other scaffold proteins to autism has been proposed, but results only allow to suggest that they contribute to an increased susceptibility to autism spectrum disorders.
2.3. Neuroinflammatory Diseases and Glial Cells
Neuroinflammatory diseases are a group of conditions that involve inflammation in the central nervous system (CNS). Inflammation is the normal response of the immune system to injury or infection, but in some cases, it can become chronic and contribute to the development of various neurological disorders. Neuroinflammatory diseases include, among others, multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS), and also those derived from a brain trauma or brain stroke. Moreover, glial cell activation or alterations contribute significantly to this group of brain diseases.
Several studies have highlighted structural and functional changes in the AIS after brain damage due to blast wave, oxygen/glucose deprivation, or brain ischemia. Exposure to a blast wave generates a traumatic brain injury, leading to altered cognition, memory, and behavior. Translated to a rat model, the consequences are a shortening of the AIS and decreased neuronal excitability [
112]. While the cellular and molecular changes are unknown, other models of brain injury have allowed us to understand several potential mechanisms. An oxygen and glucose deprivation model representative of brain ischemia allowed us to determine that sodium channels, structural proteins, and membrane proteins are proteolyzed by calpain [
16]. As mentioned above, one of the mechanisms that activates calpain in the AIS is mediated by the purinergic P2X7 receptor. This receptor is not located in the AIS but its inhibition protects AIS voltage-gated ion channels and AIS structural proteins protecting the AIS integrity after middle cerebral artery occlusion (MCAO)-induced ischemia [
12].
ALS and MS are characterized by alterations in nerve impulse conduction. In addition to myelin sheath alterations, models of both diseases show alterations in voltage-gated ion channel expression in the AIS and AIS plasticity. Studies have shown that the expression of Na
v1.6 channels is increased in demyelinated axons in MS patients, activating the accumulation of intra-axonal calcium through the Na
+/Ca
2+ exchanger, which may contribute to the development of axonal dysfunction and neurodegeneration [
113]. Motoneuron AISs in an ALS mice model G127X SOD1 were longer and thinner during the symptomatic stage and results suggest that the Na
v1.6 channel expression increases [
114]. However, G127X SOD1 mice show shorter AISs before the symptomatic stage, suggesting that AIS plasticity mechanisms may be a target for ALS. Other ALS models show reduced selective transport of cargoes before pathology appeared, suggesting AIS dysfunction [
115].
A recent study on multiple sclerosis human tissue has shown that the gap between soma and AIS increases in pyramidal neurons and Purkinje cells in multiple sclerosis tissue and that the AIS is longer in cortical neurons of cortical inactive lesions [
116]. Whether these AIS plasticity changes lead to voltage-gated ion channel expression or function alterations remains elusive, and further studies are necessary to decipher the potential role of the AIS in ALS and MS.
Glial cells play an important role in brain physiology and neuronal modulation. Astrocytes and microglia play a role in ischemia, ALS, or MS; however, their potential relation with the AIS needs further investigation. A small percentage of AISs in the cortex are associated and contact with microglial cells, and interestingly activated microglia lost its interaction with the AIS [
117]. This interaction seems to have a physiological role and needs an intact AIS to be preserved. The role of this microglia–AIS contact in the regulation of voltage-gated ion channels is unknown; however, it is essential for chandelier cell GABAergic synaptogenesis in the AIS of neocortical pyramidal neurons [
118].
Numerous studies point towards a crucial role of astrocytic ion channels linking the CNS microenvironment to astroglial responses, modulating health, inflammation, and ischemia. Even if astrocytes are non-excitable cells, these cells express a battery of potassium and calcium voltage-gated ion channels, as well as other ion channels that act as sensors and modulators [
119]. Increased intracellular Ca
2+ promotes ATP release from astrocytes, which modulates neuronal excitability and decreases the conduction speed in myelinated axons through their action on A2aR receptors and HCN2 channels in the AIS and nodes of Ranvier [
120].
The role of microglia and astrocytes in brain diseases related to neuronal excitability and survival is increasingly studied; however, how these cells can participate in disease onset regulating their own ion channels and those in neurons remains unknown.
2.4. Autoimmune Diseases
A group of autoimmune diseases have been characterized as affecting structural and functional proteins in the AIS. Autoantibodies to βIV spectrin and TRIM46 have been detected in paraneoplastic CNS disorders [
121,
122,
123]. AnkyrinG autoantibodies have been identified in some people affected by viral infections or suffering from cancer. AnkyrinG autoantibodies have been identified in AIS, nodes of Ranvier, and the subpial space in a patient living with human immunodeficiency virus (HIV) suffering steroid-responsive meningoencephalitis [
124] and in a patient with a diagnosis of metastatic ovarian cancer and seizures [
124]. Autoantibodies to Lgi1 are characteristic of limbic encephalitis [
125]. Lgi1 interacts with the K
v1.1 channel and is required to modulate the K
v1.1 density in the AIS. Lgi1 knockout mice show increased neuronal excitability and suffered from seizures [
126]. Together with Lgi1, Caspr2 also interacts with the K
v1.1 channel, and the appearance of Caspr2 autoantibodies is associated with autoimmune encephalitis [
127]. Autoantibodies to voltage potassium channels are also related to Morvan’s syndrome and have been found in schizophrenia patients [
128].
2.5. Alzheimer’s Disease
Recent studies have proposed an excitatory/inhibitory imbalance as a potential onset mechanism for Alzheimer’s disease. Actually, voltage-gated sodium channels and ankyrinG expression was significantly decreased in an Alzheimer’s disease mouse model APP/PS1 [
129] from postnatal stages. Young APP/PS1 mice showed a reduced number of action potentials, while older APP/PS1 mice have an increased number of spikes and hyperexcitability [
130]. This maybe the consequence of an increased number of cell surface Na
v1.6 channels at around 7 months [
131]. In fact, Na
v1.6 knockdown is capable of ameliorating cognitive defects and reducing hyperexcitability in APP/PS1 mice [
132]. Meanwhile, there are no data regarding K
v channel expression at the AIS in Alzheimer’s mice models, even though interaction of K
v7 channels with BACE1 was described [
133]. Altered calcium homeostasis in neurons or glial cells is an early event observed in mouse models of Alzheimer disease [
134]. Regarding the AIS, the potential role of the cisternal organelle in Ca
2+ homeostasis deregulation has not been studied; however, the presence of ryanodine receptors coupled to Ca
v3 channels in the AIS [
55] suggests the role of these channels in action potential deregulation.
Moreover, AIS shortening was identified in Alzheimer’s disease mouse models or patients [
135,
136,
137]. The absence of AIS structural plasticity was demonstrated in a frontotemporal dementia model due to a tau protein mutation identified in humans and its relation to EB1/EB3 proteins and microtubules in the AIS [
138].
In conclusion, the dysregulation of voltage-gated ion channels can have profound effects on neuronal function and contribute to the development of mental disorders and neurodegenerative diseases. Understanding the role of these channels in disease pathogenesis may lead to the development of new treatments and therapies for these devastating conditions. However, it is important to take into account that voltage-gated ion channels pathologies are not only due to alterations in channel structure or expression, but also due to changes in the neuronal domain where their function is exerted and changes in their scaffold and interacting proteins.