Congenital adrenal hyperplasia (CAH) is a common genetic disorder in endocrinology, especially its milder clinical presentation, often caused by a partial or total deficiency of the 21-hydroxylase enzyme located in the adrenal cortex. CAH is characterized by the overproduction of androgen, along with variable degrees of cortisol and aldosterone deficiency. The age at diagnosis can provide some information about underlying mutations, with those diagnosed at birth/early infancy more likely to have severe enzymatic defects, which may include adrenal insufficiency, sexual development disorders, short stature in adulthood, hirsutism, and a higher risk for metabolic syndrome and infertility. Non-classic CAH, a milder form of CAH, is usually manifested later in life and is a common differential diagnosis of Polycystic Ovary Syndrome and should be actively evaluated during initial studies of clinical or biochemical hyperandrogenism.
1. Introduction
Congenital adrenal hyperplasia (CAH) is a group of genetic disorders with a wide spectrum of clinical manifestations and is, therefore, challenging in endocrine practice. More precisely, the term CAH refers to a group of genetic disorders that affect the adrenal glands. These are autosomal recessive alterations that cause metabolic dysregulations, including impaired cortisol synthesis, due to deficient activity of enzymes located in the zona fasciculata of the adrenal cortex. In most cases (>95%), these are mutations that affect the CYP21A2 gene, which encodes the enzyme 21-hydroxylase. Although all forms of CAH reduce cortisol production, this may occur at different degrees and various levels, affecting distinct segments of the adrenal steroid biosynthesis pathway.
2. Epidemiology
Although its incidence depends on ethnic (inbred) and geographic factors, classic CAH (caused by D21OH) is a rare disease with a prevalence that ranges from 1 in 10,000 to 1 in 16,000 cases
[1]. In contrast, non-classic CAH (NCCAH; also called late-onset CAH) has a much higher frequency (up to 1/500); NCCAH is also common within ethnic groups with high consanguinity. Worldwide, CAH (including mild cases) is considered the most common autosomal recessive disease, surpassing cystic fibrosis and phenylketonuria
[2].
3. Pathophysiology and Classification of Congenital Adrenal Hyperplasia Due to 21-Hydrolase Deficiency
As explained, alterations in the CYP21A2 gene translate into enzymatic deficiency of the 21-hydroxylase (21OHD) activity causing a decrease in cortisol biosynthesis. However, in certain severe cases, CYP21A2 mutations can also affect aldosterone production. Then, the lack of cortisol feedback results in increased ACTH, which promotes the accumulation of 17-hydroxyprogesterone (17OHP) and other steroids that serve as substrates for androgen excess. Allelic CYP21A2 variants are associated with a continuum spectrum of enzyme phenotypes. As pointed out, CAH is a recessive genetic disorder. Therefore, affected subjects can be either homozygous (same mutation affects both alleles) or compound heterozygous (two different mutations affecting each allele). Importantly, the onset and severity of the clinical manifestations of the disease will ultimately depend on the allele affected by the “mildest” mutation
[3]. Clinical CAH phenotypes are characterized by decreased cortisol synthesis and increased androgen secretion and depend on both the age at presentation and the severity of the CYP21A2 mutation. Hence, CAH cases can be divided into three categories
[1]: (a) Salt-wasting (SW) represents 65–75% of the classic CAH cases. These are manifested in infancy and are characterized by a marked cortisol and aldosterone deficiency along with hyperandrogenism. Residual enzymatic activity of SW-CAH is typically < 1%. (b) Simple virilizing CAH comprises 25–35% of the classic CAH. Unlike SW-CAH, this form manifests later in life and is characterized by a severe cortisol deficit, but unaltered aldosterone. Residual enzymatic activity of this form of CAH is 1–2%; and (c) Non-classic CAH is the most frequently seen in the clinic. It usually manifests during puberty and is characterized by hyperandrogenism. Residual enzymatic activity ranges from 20% to 50%
[4] (
Figure 1) and normal cortisol production is maintained by excess ACTH.
Figure 1. Deregulated hypothalamic–pituitary–adrenal axis function in congenital adrenal hyperplasia due to 21-hydroxylase deficiency.
As noted, CAH subtypes are determined by the severity of underlying mutation(s). For example, a heterozygous individual that harbors a “severe” and a “mild” mutation will fall into the NCCAH subtype. Therefore, an individual that harbors a “severe” mutation but also carries a wild-type allele will not manifest the disease
[5] (
Figure 2).
Figure 2. Most frequent pathogenic variants in adrenal hyperplasia due to 21-hydroxylase deficiency.
4. Clinical Features
4.1. SW Classic CAH
Severe aldosterone and cortisol deficiency in this condition can lead to a salt-wasting crisis in newborns. However, this is unlikely to occur within 5 days of birth, which is usually when newborns are discharged from the hospital. Therefore, SW-CAH requires high diagnostic suspicion, particularly among males due to the lack of genital ambiguity. Affected individuals typically present with adrenal insufficiency associated with hypovolemic shock, hyponatremia, hyperkalemia, metabolic acidosis, and sometimes hypoglycemia. In 46, XX individuals, this condition is accompanied by various degrees of virilization due to exposure to excess androgens during intrauterine development. Although the uterus, fallopian tubes, and ovaries are formed normally, XX DSD may involve clitoral hypertrophy, labial fusion, and urogenital sinus defects. The Prader classification ranges from 1 to 5, where grade 1 displays mild clitoral hypertrophy and grade 5 is characterized by complete masculinization. Excess adrenal androgens do not affect 46, XY sexual differentiation.
4.2. Simple Virilizing CAH
This condition is usually detected before puberty and evidenced by clinical signs, such as hyperandrogenism, premature puberty/adrenarche, apocrine odor, precocious puberty, clitoromegaly, rapid growth, and accelerated skeletal maturation (with a compromise of final height). Most patients display subclinical cortisol deficiency but preserve their mineralocorticoid function
[1][6].
4.3. Non-Classical CAH
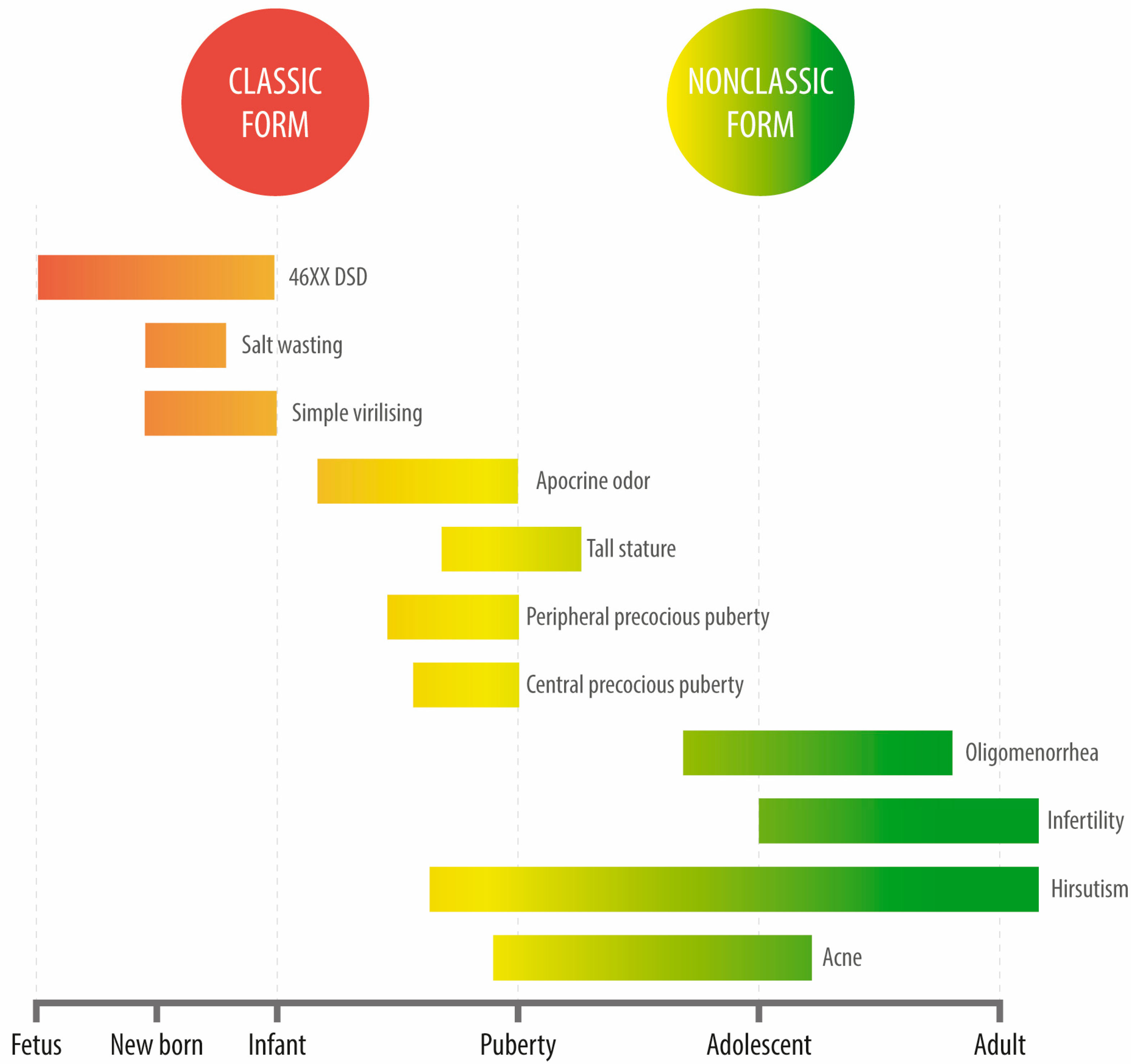
Unlike classic CAH, cortisol and aldosterone levels in these patients remain unaffected. The most frequent reason for consultation among these patients is late-onset hyperandrogenism, generally manifested either in the peri-pubertal stage or during adulthood. This condition is severely underdiagnosed among males given the evident difficulties in assessing hyperandrogenism. During adolescence, the most frequent reasons for consultation are acne, hirsutism, or oligomenorrhea (a condition that is clinically indistinguishable from polycystic ovary syndrome (PCOS)). Affected individuals may also display polycystic ovarian morphology. Oftentimes, NCCAH diagnosis occurs later in life, even into adulthood, in the context of infertility studies or after recurrent miscarriages (Figure 3).
Figure 3. Clinical manifestations of congenital adrenal hyperplasia due to 21-hydroxylase deficiency at different ages.
5. Treatment of CAH
5.1. Classic CAH
The treatment of classical CAH involves achieving a balance of three key clinical goals: to promote the physiological replacement of adrenal insufficiency, to reduce the exposure to age- and sex-appropriate levels of adrenal androgens, and to avoid iatrogenic hypercortisolism and its potentially associated comorbidities
[7].
Glucocorticoid replacement clinical recommendations: Hydrocortisone is typically the preferred choice and should be administered two or three times per day, with higher morning and lower evening doses. Children usually require 10–15 mg/m2 per day every 8 h, while adults may require 15–25 mg per day with a last dose administered at least 6 h before bedtime to maintain the circadian rhythm.
In newborn patients suspected of having the salt-wasting (SW) form of CAH due to high levels of 17OHP in the screening sample, urgent clinical advice is necessary for appropriate treatment. The initial dose of hydrocortisone given to neonates and infants with a newly detected SW form should be adjusted based on their clinical situation. If the newborn has elevated potassium and decreased sodium levels, prompt treatment should begin, including intravenous glucose infusion containing sodium and intravenous hydrocortisone. The initial hydrocortisone bolus dose suggested is 5 mg/kg, followed by 25 mg per 24 h as a continuous infusion or divided into three or four doses. As children grow older, the recommended hydrocortisone dose for CAH typically increases. Children may require a daily dose of 10 mg/m
2 every 8 h. For adults with CAH, the recommended dose may range from 15 to 25 mg per day, with the last dose administered at least 6 h before bedtime to maintain the circadian rhythm
[8][9].
In cases where ACTH remains excessively elevated despite treatment, a low dose of nocturnal prednisone (1–2 mg) can be prescribed, especially after puberty
[7]. In cases where a patient is struggling with adherence to evening hydrocortisone or experiencing hyperpigmentation, the alternative is a single morning dose (5–7.5 mg/d) of long half-life glucocorticoids, such as prednisone. In contrast, dexamethasone (0.25–0.5 mg/d dose) is not recommended given the difficulty of titration and the risk of an iatrogenic Cushing’s syndrome
[4]. It is noteworthy that monotherapy with nocturnal corticosteroids is not appropriate in these cases as they can lead to long-term sleep and/or mood disorders. Of note, long-acting glucocorticoids could lead to detrimental effects on the growth of children with severe forms of CAH
[10][11]. In children, the growth velocity, weight, blood pressure, bone age, signs of hyperandrogenism, and the presence of adrenal remnants in the testes should be closely monitored by ultrasound
[4][7].
Mineralocorticoid replacement clinical recommendations: Fludrocortisone is often the drug of choice.
In rare cases where medical management cannot effectively control hyperandrogenism, or in cases where its control would require producing iatrogenic hypercortisolism, bilateral adrenalectomy may be considered. This option is common for adenomas and large bilateral myelolipomas secondary to ACTH stimulation
[12].
5.1.1. Treatment under Medical-Surgical Stress Conditions
Classic CAH patients are at a high risk of suffering adrenal insufficiency crises manifested as hypoglycemia, seizures, hyponatremia, hyperkalemia, dehydration, and/or refractory shock. To prevent these potentially life-threatening events, all the affected patients should be informed about these risks and carry identification in the form of a bracelet or a badge. Patients should also maintain emergency corticosteroids at hand in case they are unable to take their regular oral medication due to complications, such as vomiting or severe diarrhea. Notably, certain situations, such as fever (>38.5 °C), tooth extraction, simple outpatient surgery, or endoscopic procedures can increase the risk of adrenal insufficiency crises. For these cases, the recommendation is to double the usual corticosteroid dose to prevent these events
[13].
5.1.2. New Drugs to Improve the Treatment of CAH Patients
Recent research has sought to replicate the circadian rhythm of glucocorticoid supplementation to reduce exposure to these compounds. Modified or delayed-release hydrocortisone formulations and continuous subcutaneous delivery of hydrocortisone may be useful for patients with a rapid cortisol metabolism or impaired gut absorption. However, the long-term clinical utility beyond improved biochemical parameters is still uncertain
[14]. In pre-pubertal CAH patients, medications that reduce androgen production and/or its action, such as testolactone, flutamide, and abiraterone acetate, may be useful for androgen suppression until the anticipated age of puberty. However, further studies are necessary to confirm their effectiveness and safety. Lastly, CRH antagonists have recently emerged as promissory treatments in patients with poor disease control CAH
[15].
5.1.3. Restoring Functional Anatomy in 46, XX Subjects Virilized by Classic CAH
For 46, XX virilized individuals with classic CAH, surgery is not a medical emergency unless there are urogenital sinus complications. These cases should seek treatment exclusively at referral centers with pre-established protocols and a multidisciplinary team of professionals specialized in DSD management
[4].
5.1.4. Preventing Prenatal Virilization in At-Risk Pregnancies
For >40 years, dexamethasone has been proposed as a treatment to prevent the virilization of a female fetus that inherits two severely mutated CYP21A2 alleles from her mother with classic CAH during pregnancy. Although the main goal of dexamethasone is to suppress fetal ACTH production, its use can result in pharmacological hypercortisolism, affecting both the mother and the fetus since dexamethasone is not inactivated in the placenta. Studies recommend a preconception dose of 20 ug/kg and up to 1.5 mg which is 2–6 times the maternal physiological requirement and almost 60 times that of the fetus. Although this intervention is successful in 85% of cases, effectively reducing the Prader score by about 40%
[16], it is important to weigh up its risks and benefits. Some of these include teratogenicity in animals (class C drug) and maternal Cushing’s syndrome
[17]. Importantly, it should be noted that seven out of eight fetuses will be treated unnecessarily, at least until the biological sex of the individual is known. Furthermore, given the effectiveness of genital surgery, current international guidelines do not recommend routine prenatal dexamethasone treatments. This intervention should be considered rather as an experimental procedure performed at tertiary centers after obtaining informed consent signed by both parents and the approval of an ethics committee
[4]. The current recommendation to avoid the unnecessary treatment of male fetuses is to perform early measurements of the fetal Y chromosome in maternal blood, starting at 6–8 weeks of gestation.
5.2. NCCAH
By definition, NCCAH does not involve adrenal insufficiency. Therefore, in most cases, cortisol replacement is neither necessary nor recommended, especially in adult individuals. Despite this, the current guidelines from the Endocrine Society recommend cortisol replacement in two specific cases: (1) in adult women with hyperandrogenism and fertility disorders and (2) in pediatric patients with rapidly progressive precocious puberty associated with advanced bone age. Nonetheless, in patients with central precocious puberty, it is possible to halt puberty at the hypothalamic level using GnRH agonists, avoiding the use of corticosteroids and their potential adverse effects
[4].
As mentioned earlier, NCCAH often affects adolescent or adult women with acne and hirsutism. As with PCOS, the typical first-line treatment is a combination of oral contraceptives. In patients that display a poor response to treatment, antiandrogens, such as spironolactone (initial dose 50–100 mg/day) or low doses of flutamide (62.5–125 mg/day), may be added to the treatment regimen
[4].
 +1 credit
+1 credit