Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yichun Zhang | -- | 1240 | 2023-04-25 05:28:41 | | | |
| 2 | Dean Liu | + 246 word(s) | 1486 | 2023-05-04 02:21:54 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Li, S.; Zhang, Y.; Li, K.; Liu, Y.; Chi, S.; Wang, Y.; Tang, S. Hirschsprung-Associated Enterocolitis. Encyclopedia. Available online: https://encyclopedia.pub/entry/43423 (accessed on 24 July 2026).
Li S, Zhang Y, Li K, Liu Y, Chi S, Wang Y, et al. Hirschsprung-Associated Enterocolitis. Encyclopedia. Available at: https://encyclopedia.pub/entry/43423. Accessed July 24, 2026.
Li, Shuai, Yichun Zhang, Kang Li, Yuan Liu, Shuiqing Chi, Yong Wang, Shaotao Tang. "Hirschsprung-Associated Enterocolitis" Encyclopedia, https://encyclopedia.pub/entry/43423 (accessed July 24, 2026).
Li, S., Zhang, Y., Li, K., Liu, Y., Chi, S., Wang, Y., & Tang, S. (2023, April 25). Hirschsprung-Associated Enterocolitis. In Encyclopedia. https://encyclopedia.pub/entry/43423
Li, Shuai, et al. "Hirschsprung-Associated Enterocolitis." Encyclopedia. Web. 25 April, 2023.
Copy Citation
Despite the significant progress that has been made in terms of understanding the pathophysiology and risk factors of Hirschsprung-associated enterocolitis (HAEC), the morbidity rate has remained unsatisfactorily stable, and clinical management of the condition continues to be challenging.
Hirschsprung disease (HSCR)
enterocolitis
Hirschsprung-associated enterocolitis (HAEC)
1. Introduction
Hirschsprung-associated enterocolitis (HAEC) is the leading cause of serious morbidity and mortality in patients with Hirschsprung disease (HSCR) [1]. Clinically, among the various risk factors, delayed diagnosis, the type of operation employed, female sex, having a younger age at presentation, long-segment disease, family history, associated anomalies, and anastomotic leaks or strictures are the most commonly reported in the literature [2]. One of the leading causes or risk factors for HAEC has been considered to be partial mechanical obstruction. The major underlying feature of this may be anastomotic stricture, bowel-disordered motility, or functional obstruction (paralysis). There is some evidence to suggest that recurrent HAEC may be released following internal sphincter myotomy [3]. However, HAEC also occurs in patients with enterostomy, and without any evidence of obstruction. Therefore, other factors must be involved.
Given the numerous advances that have been made in terms of performing meticulous operations procedures, standardized diagnostic systems [4], and postoperative management [5], the reported incidence of HAEC remains stable, ranging from 6 to 60% prior to pull-through surgery, and from 25 to 37% following surgery [4]. Several studies have found that the proximal dilated segment of the colon is mostly affected and more susceptible to HAEC [6][7][8], which seems to explain part of the reason why HAEC still occurs postoperatively. In addition, some studies have found that older age at radical surgery is a risk factor for the development of postoperative HAEC [9], and older age is also a risk factor for the development of preoperative HAEC [10]. However, the data concerning HAEC etiology remain limited.
On the other hand, since the comprehensive review by Demehri [11], basic research on HAEC has made marked progress in terms of increasing the knowledge base, especially with respect to studies on genes, the microbiome, immunity, and other aspects [12][13].
2. Genes
Previous studies have revealed that genetic background influences HAEC, mainly in the form of changes in the incidence and severity of HAEC in patients with HSCR with a combination of several clinical syndromes. Kwendakwema [14] conducted a retrospective cohort study of 207 patients with HSCR, 26 (13%) of whom were trisomy 21 (T21) patients, and found that the incidence of HAEC in children with HSCR and T21 (38%) was not significantly different from that in children with HSCR alone (41%). By contrast, Halleran’s study found that, compared with patients with HAEC alone, patients with a combination of HAEC and T21 experienced more severe symptoms, including longer duration of symptoms, hypotension, greater likelihood of tachycardia and longer times on antibiotics, and were also more likely to require intensive care unit admission [15]. A higher incidence of HAEC in children with Mowat-Wilson syndrome with HSCR has also been reported [16].
The advent of whole-exome sequencing has also facilitated this type of research. Bachetti [17] identified p.H187Q in the oncostatin-M (OSM) receptor (OSMR) gene as a susceptibility variant of HAEC. It may exert a key role in HAEC pathogenesis by regulating/activating the OSM-OSMR axis. Via large-sample sequencing, the single nucleotide polymorphisms (SNPs) rs8104023 [18] and rs2191026 [19] were found to be significantly associated with postoperative HAEC. In another study, DNA was extracted from the colon tissue samples of 30 patients with HAEC, and the mRNA expression of integrin beta-2 (ITGB2; also known as CD18) was found to be negatively correlated with the incidence and severity of HAEC [20].
Taken together, the above studies have suggested that the pathogenesis of HAEC is closely associated with the underlying genes and the genetic background (Figure 2), although the specific mechanism(s) involved still require further study.
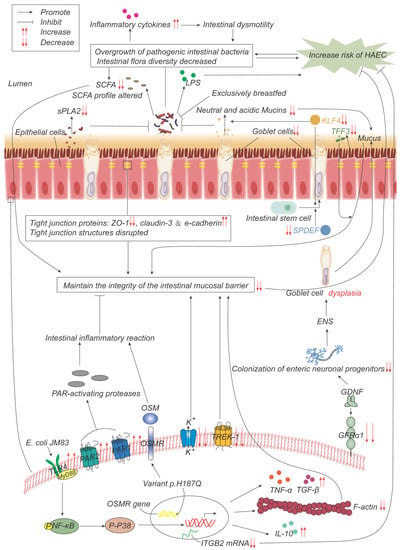
Figure 2. Pathogenesis advances in genes, intestinal microflora, and mucosal barrier. p.H187Q in the Oncostatin-M receptor (OSMR) gene is a susceptible variant of HAEC, which promotes inflammation by activating the OSM-OSMR axis. Decreased integrin beta-2 (ITGB2) mRNA expression is negatively correlated with the incidence and severity of HAEC. Overgrowth and reduced diversity of intestinal bacteria lead to increased release of inflammatory factors, which leads to intestinal dysmotility, which in turn leads to further bacterial overgrowth. Epithelial cells produce less secretory phospholipase A2 (sPLA2), which reduces the inhibition of bacteria and promotes bacterial overgrowth. Lipopolysaccharide (LPS) produced by bacteria can promote the development of HAEC, while exclusive breastfeeding regulates the gut microbiome in such a way that LPS production is reduced. Short-chain fatty acids (SCFAs) produced by bacteria are reduced, and their composition is altered, impairing the function of maintaining mucosal integrity. The expressions of TFF3, SPDEF, and KLF4 are significantly down-regulated, leading to the decrease in goblet cells (GCs) and the secretion of neutral and acidic mucins, which lead to the weakening of the mucosal barrier function. The expression of glial cell line-derived neurotrophic factor (GDNF) co-receptor and GDNF family receptor alpha-1 (GFRα1) is decreased, and the colonization of neuronal progenitor cells in the intestine is impaired, affecting ENS development, resulting in GCs dysplasia, and abnormal mucin production and storage. The structure of the tight-junction protein is damaged, and its composition is changed, which impairs its function of maintaining the mucosal barrier integrity. Escherichia coli JM83 stimulates NF-κB through TLR4 and MyD88, and through NF-κB/p-p38 signal transduction, F-actin protein density is significantly reduced, IL-10, TNF-α, TGF-β increase, leading to intestinal mucosal damage and promoting the development of HAEC. TREK-1 and K(ATP) channels are reduced, leading to barrier dysfunction. Increased expressions of PAR-1 and PAR-2 lead to excessive local release of PAR-activating protease, which leads to inflammatory responses and impairs barrier function.
3. Intestinal Microbiome
The human intestinal microbiome is a complex ecosystem in which the phyla Firmicutes and Bacteroidetes are dominant, followed by proteobacteria [21], and the normal intestinal flora is in a dynamic balance. Maintaining the relative balance of the intestinal microflora is closely associated with the stability of the internal environment of the intestine and its normal function.
4. Intestinal Mucosal Barrier
The mucosal barrier serves as the first line of defense, protecting the healthy intestinal surface from adhesion and invasion by tubular microorganisms. The components of the intestinal barrier include the lumen, microenvironment or mucus-containing layer, epithelium, and lamina propria. Numerous studies have shown that structural defects and dysfunction of the intestinal mucosal barrier are responsible for the pathogenesis of HAEC.
5. Enteric Nervous System (ENS)
The abnormal ENS of HSCR leads to impaired intestinal motility, resulting in functional obstruction with subsequent stasis and overgrowth of pathogenic intestinal bacteria, destruction of the mucosal layer, invasion of the intestinal wall, dysfunction of the intestinal mucosal barrier, impaired immune response and consequent HAEC. This abnormal neurological function is considered to be an important cause of preoperative HAEC episodes.
6. Immune System
Intestinal-associated lymphoid tissue is the largest immune organ in the body. Macrophages that have colonized the intestine are the first line of defense of the intestinal immune response, and serve to protect the intestine from pathogenic microorganisms. B lymphocytes mature and differentiate into plasma cells that secrete secretory immunoglobulin A (sIgA) into the intestinal lumen, which is the main immunoglobulin in the intestine and is actively transported to the mucosal surface as a dimer through the polymeric immunoglobulin receptor (pIgR), thereby maintaining the balance and normal function of the intestinal microenvironment [22]. There is a close relationship between the immune system and HAECs. For example, immune organs, immune cells, immunoglobulin, cytokines, inflammasome and exosomes are involved in the occurrence of HaECs (Figure 4).
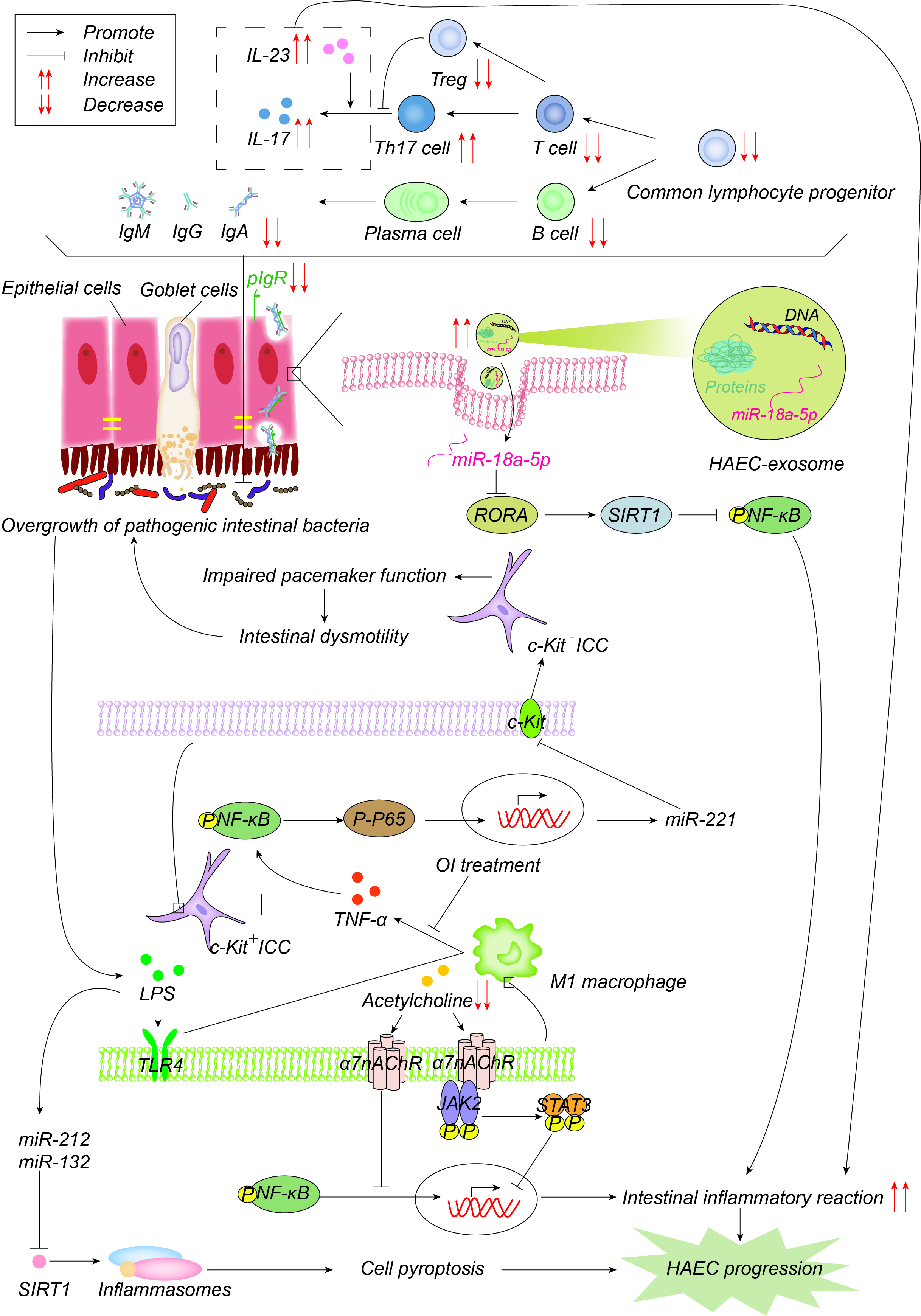
Figure 4. Pathogenesis advances in immunity. The number of common lymphocyte progenitor populations and, therefore, T cells and B cells decrease significantly, resulting in reduced immunoglobulin, which is actively transported to the mucosal surface by polymerized immunoglobulin receptors (pIgR) in the form of dimers to maintain the intestinal microenvironment balance and normal function. pIgR is also reduced, which together leads to intestinal microenvironment disturbances and bacterial overgrowth. Increased Th17 cells produce more IL-17 and promote the intestinal inflammatory response. On one hand, LPS produced by bacteria activates M1 macrophages through the TLR4 pathway, and M1 macrophages produce TNF-α, which inhibits c-Kit expression in ICC through the NF-κB/miR-221 pathway, leading to intestinal motility disorders, which further leads to the accumulation and overgrowth of intestinal bacteria. 4-octyl itaconate (OI) reduces the production of proinflammatory factors and promotes ICC phenotypic recovery by inhibiting macrophage activation. LPS, on the other hand, induces the up-regulation of miR-132/-212, activates inflammasome NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) by inhibiting the expression of sirtuin 1 (Sirt1), promotes cell pyroptosis, and then promotes the occurrence and development of HAEC. Acetylcholine acts on α7 nicotinic acetylcholine receptor (α7nAChR) on the surface of macrophages, inhibits the activation of macrophages, activates the anti-inflammatory pathway of JAK2-STAT3 and suppresses the inflammatory pathway of NF-κB. Acetylcholine in HAEC is reduced, and the above anti-inflammatory effects are weakened. Exosome miR-18a-5p down-regulates RAR-related orphan receptor A (RORA), activates the SIRT1/NF-κB signaling pathway, induces excessive inflammatory response, and thus promotes the development of HAEC.
References
- Tam, P.K.H.; Chung, P.H.Y.; St Peter, S.D.; Gayer, C.P.; Ford, H.R.; Tam, G.C.H.; Wong, K.K.Y.; Pakarinen, M.P.; Davenport, M. Advances in paediatric gastroenterology. Lancet 2017, 390, 1072–1082.
- Le-Nguyen, A.; Righini-Grunder, F.; Piche, N.; Faure, C.; Aspirot, A. Factors influencing the incidence of Hirschsprung associated enterocolitis (HAEC). J. Pediatr. Surg. 2019, 54, 959–963.
- Zhang, J.; Li, L.; Hou, W.; Liu, S.; Diao, M.; Zhang, J.; Ming, A.; Cheng, W. Transanal rectal mucosectomy and partial internal anal sphincterectomy for Hirschsprung’s disease. J. Pediatr. Surg. 2014, 49, 831–834.
- Gosain, A.; Frykman, P.K.; Cowles, R.A.; Horton, J.; Levitt, M.; Rothstein, D.H.; Langer, J.C.; Goldstein, A.M. Guidelines for the diagnosis and management of Hirschsprung-associated enterocolitis. Pediatr. Surg. Int. 2017, 33, 517–521.
- Langer, J.C.; Rollins, M.D.; Levitt, M.; Gosain, A.; de la Torre, L.; Kapur, R.P.; Cowles, R.A.; Horton, J.; Rothstein, D.H.; Goldstein, A.M.; et al. Guidelines for the management of postoperative obstructive symptoms in children with Hirschsprung disease. Pediatr. Surg. Int. 2017, 33, 523–526.
- Chen, X.; Meng, X.; Zhang, H.; Feng, C.; Wang, B.; Li, N.; Abdullahi, K.M.; Wu, X.; Yang, J.; Li, Z.; et al. Intestinal proinflammatory macrophages induce a phenotypic switch in interstitial cells of Cajal. J. Clin. Investig. 2020, 130, 6443–6456.
- Chen, F.; Wei, X.; Chen, X.; Xiang, L.; Meng, X.; Feng, J. Role of Janus kinase2-signal transducer and activator of transcription3 signal transduction pathway in hirschsprung’s disease associated enterocolitis. Chin. J. Exp. Surg. 2020, 37, 1089–1092.
- Chen, F.; Wei, X.; Chen, X.; Xiang, L.; Meng, X.; Feng, J. Role of NF-kappaB signal transduction pathway in Hirschsprung’s disease associated enterocolitis. Chin. J. Pediatr. Surg. 2020, 41, 1123–1127.
- Sakurai, T.; Tanaka, H.; Endo, N. Predictive factors for the development of postoperative Hirschsprung-associated enterocolitis in children operated during infancy. Pediatr. Surg. Int. 2021, 37, 275–280.
- Yulianda, D.; Sati, A.I.; Makhmudi, A.; Gunadi. Risk factors of preoperative Hirschsprung-associated enterocolitis. BMC Proc. 2019, 13, 18.
- Demehri, F.R.; Halaweish, I.F.; Coran, A.G.; Teitelbaum, D.H. Hirschsprung-associated enterocolitis: Pathogenesis, treatment and prevention. Pediatr. Surg. Int. 2013, 29, 873–881.
- Tilghman, J.M.; Ling, A.Y.; Turner, T.N.; Sosa, M.X.; Krumm, N.; Chatterjee, S.; Kapoor, A.; Coe, B.P.; Nguyen, K.H.; Gupta, N.; et al. Molecular Genetic Anatomy and Risk Profile of Hirschsprung’s Disease. N. Engl. J. Med. 2019, 380, 1421–1432.
- Tam, P.; Wong, K.; Atala, A.; Giobbe, G.G.; Booth, C.; Gruber, P.J.; Monone, M.; Rafii, S.; Rando, T.A.; Vacanti, J.; et al. Regenerative medicine: Postnatal approaches. Lancet Child Adolesc. 2022, 6, 654–666.
- Kwendakwema, N.; Al-Dulaimi, R.; Presson, A.P.; Zobell, S.; Stevens, A.M.; Bucher, B.T.; Barnhart, D.C.; Rollins, M.D. Enterocolitis and bowel function in children with Hirschsprung disease and trisomy 21. J. Pediatr. Surg. 2016, 51, 2001–2004.
- Halleran, D.R.; Ahmad, H.; Maloof, E.; Paradiso, M.; Lehmkuhl, H.; Minneci, P.C.; Levitt, M.A.; Wood, R.J. Does Hirschsprung-Associated Enterocolitis Differ in Children With and Without Down Syndrome? J. Surg. Res. 2020, 245, 564–568.
- Li, Q.; Zhang, Z.; Li, L.; Jiang, Q. Diagnosis, treatment and prognosis of Mowat-Wilson syndrome with Hirschsprung’s disease. Chin. J. Pediatr. Surg. 2022, 43, 522–527.
- Bachetti, T.; Rosamilia, F.; Bartolucci, M.; Santamaria, G.; Mosconi, M.; Sartori, S.; De Filippo, M.R.; Di Duca, M.; Obino, V.; Avanzini, S.; et al. The OSMR Gene Is Involved in Hirschsprung Associated Enterocolitis Susceptibility through an Altered Downstream Signaling. Int. J. Mol. Sci. 2021, 22, 3831.
- Zhang, H.; Zhao, J.L.; Zheng, Y.; Xie, X.L.; Huang, L.H.; Li, L.; Zhu, Y.; Lu, L.F.; Hu, T.Q.; Zhong, W.; et al. Correlation analysis of IL-11 polymorphisms and Hirschsprung disease subtype susceptibility in Southern Chinese Children. BMC Med. Genom. 2021, 14, 21.
- Xie, X.; He, Q.; Huang, L.; Li, L.; Yao, Y.; Xia, H.; Zhao, J.; Zhong, W.; Zhang, Y. Associations of SLC6A20 genetic polymorphisms with Hirschsprung’s disease in a Southern Chinese population. Biosci. Rep. 2019, 39, BSR20182290.
- Mariana, N.; Islam, A.A.; Massi, M.N.; Warsinggih; Hatta, M.; Patellongi, I. ITGB2 (CD18) mRNA expression in hirschsprung-associated enterocolitis (HAEC). Indian J. Public Health Res. Dev. 2019, 10, 1015–1020.
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180.
- Turula, H.; Wobus, C.E. The Role of the Polymeric Immunoglobulin Receptor and Secretory Immunoglobulins during Mucosal Infection and Immunity. Viruses 2018, 10, 237.
More
Information
Subjects:
Gastroenterology & Hepatology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
901
Revisions:
2 times
(View History)
Update Date:
04 May 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No