Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Gastroenterology & Hepatology
尽管在了解先天性巨结肠相关性小肠结肠炎(HAEC)的病理生理学和危险因素方面取得了重大进展,但发病率仍不稳定,该病的临床管理仍然具有挑战性。
- Hirschsprung disease (HSCR)
- enterocolitis
- Hirschsprung-associated enterocolitis (HAEC)
1. Introduction
Hirschsprung-associated enterocolitis (HAEC) is the leading cause of serious morbidity and mortality in patients with Hirschsprung disease (HSCR) [1]. Clinically, among the various risk factors, delayed diagnosis, the type of operation employed, female sex, having a younger age at presentation, long-segment disease, family history, associated anomalies, and anastomotic leaks or strictures are the most commonly reported in the literature [2]. One of the leading causes or risk factors for HAEC has been considered to be partial mechanical obstruction. The major underlying feature of this may be anastomotic stricture, bowel-disordered motility, or functional obstruction (paralysis). There is some evidence to suggest that recurrent HAEC may be released following internal sphincter myotomy [3]. However, HAEC also occurs in patients with enterostomy, and without any evidence of obstruction. Therefore, other factors must be involved.
Given the numerous advances that have been made in terms of performing meticulous operations procedures, standardized diagnostic systems [4], and postoperative management [5], the reported incidence of HAEC remains stable, ranging from 6 to 60% prior to pull-through surgery, and from 25 to 37% following surgery [4]. Several studies have found that the proximal dilated segment of the colon is mostly affected and more susceptible to HAEC [6,7,8], which seems to explain part of the reason why HAEC still occurs postoperatively. In addition, some studies have found that older age at radical surgery is a risk factor for the development of postoperative HAEC [9], and older age is also a risk factor for the development of preoperative HAEC [10]. However, the data concerning HAEC etiology remain limited.
On the other hand, since the comprehensive review by Demehri [11], basic research on HAEC has made marked progress in terms of increasing the knowledge base, especially with respect to studies on genes, the microbiome, immunity, and other aspects [12,13].
2. Genes
Previous studies have revealed that genetic background influences HAEC, mainly in the form of changes in the incidence and severity of HAEC in patients with HSCR with a combination of several clinical syndromes. Kwendakwema [14] conducted a retrospective cohort study of 207 patients with HSCR, 26 (13%) of whom were trisomy 21 (T21) patients, and found that the incidence of HAEC in children with HSCR and T21 (38%) was not significantly different from that in children with HSCR alone (41%). By contrast, Halleran’s study found that, compared with patients with HAEC alone, patients with a combination of HAEC and T21 experienced more severe symptoms, including longer duration of symptoms, hypotension, greater likelihood of tachycardia and longer times on antibiotics, and were also more likely to require intensive care unit admission [15]. A higher incidence of HAEC in children with Mowat-Wilson syndrome with HSCR has also been reported [16].
The advent of whole-exome sequencing has also facilitated this type of research. Bachetti [17] identified p.H187Q in the oncostatin-M (OSM) receptor (OSMR) gene as a susceptibility variant of HAEC. It may exert a key role in HAEC pathogenesis by regulating/activating the OSM-OSMR axis. Via large-sample sequencing, the single nucleotide polymorphisms (SNPs) rs8104023 [18] and rs2191026 [19] were found to be significantly associated with postoperative HAEC. In another study, DNA was extracted from the colon tissue samples of 30 patients with HAEC, and the mRNA expression of integrin beta-2 (ITGB2; also known as CD18) was found to be negatively correlated with the incidence and severity of HAEC [20].
Taken together, the above studies have suggested that the pathogenesis of HAEC is closely associated with the underlying genes and the genetic background (Figure 2), although the specific mechanism(s) involved still require further study.
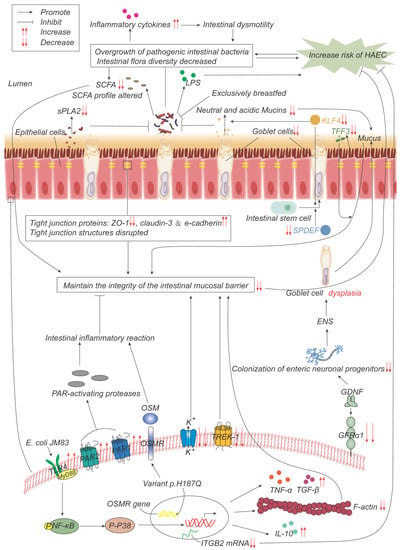
Figure 2. Pathogenesis advances in genes, intestinal microflora, and mucosal barrier. p.H187Q in the Oncostatin-M receptor (OSMR) gene is a susceptible variant of HAEC, which promotes inflammation by activating the OSM-OSMR axis. Decreased integrin beta-2 (ITGB2) mRNA expression is negatively correlated with the incidence and severity of HAEC. Overgrowth and reduced diversity of intestinal bacteria lead to increased release of inflammatory factors, which leads to intestinal dysmotility, which in turn leads to further bacterial overgrowth. Epithelial cells produce less secretory phospholipase A2 (sPLA2), which reduces the inhibition of bacteria and promotes bacterial overgrowth. Lipopolysaccharide (LPS) produced by bacteria can promote the development of HAEC, while exclusive breastfeeding regulates the gut microbiome in such a way that LPS production is reduced. Short-chain fatty acids (SCFAs) produced by bacteria are reduced, and their composition is altered, impairing the function of maintaining mucosal integrity. The expressions of TFF3, SPDEF, and KLF4 are significantly down-regulated, leading to the decrease in goblet cells (GCs) and the secretion of neutral and acidic mucins, which lead to the weakening of the mucosal barrier function. The expression of glial cell line-derived neurotrophic factor (GDNF) co-receptor and GDNF family receptor alpha-1 (GFRα1) is decreased, and the colonization of neuronal progenitor cells in the intestine is impaired, affecting ENS development, resulting in GCs dysplasia, and abnormal mucin production and storage. The structure of the tight-junction protein is damaged, and its composition is changed, which impairs its function of maintaining the mucosal barrier integrity. Escherichia coli JM83 stimulates NF-κB through TLR4 and MyD88, and through NF-κB/p-p38 signal transduction, F-actin protein density is significantly reduced, IL-10, TNF-α, TGF-β increase, leading to intestinal mucosal damage and promoting the development of HAEC. TREK-1 and K(ATP) channels are reduced, leading to barrier dysfunction. Increased expressions of PAR-1 and PAR-2 lead to excessive local release of PAR-activating protease, which leads to inflammatory responses and impairs barrier function.
3. Intestinal Microbiome
The human intestinal microbiome is a complex ecosystem in which the phyla Firmicutes and Bacteroidetes are dominant, followed by proteobacteria [21], and the normal intestinal flora is in a dynamic balance. Maintaining the relative balance of the intestinal microflora is closely associated with the stability of the internal environment of the intestine and its normal function.
4. Intestinal Mucosal Barrier
The mucosal barrier serves as the first line of defense, protecting the healthy intestinal surface from adhesion and invasion by tubular microorganisms. The components of the intestinal barrier include the lumen, microenvironment or mucus-containing layer, epithelium, and lamina propria. Numerous studies have shown that structural defects and dysfunction of the intestinal mucosal barrier are responsible for the pathogenesis of HAEC.
5. Enteric Nervous System (ENS)
The abnormal ENS of HSCR leads to impaired intestinal motility, resulting in functional obstruction with subsequent stasis and overgrowth of pathogenic intestinal bacteria, destruction of the mucosal layer, invasion of the intestinal wall, dysfunction of the intestinal mucosal barrier, impaired immune response and consequent HAEC. This abnormal neurological function is considered to be an important cause of preoperative HAEC episodes.
6. Immune System
Intestinal-associated lymphoid tissue is the largest immune organ in the body. Macrophages that have colonized the intestine are the first line of defense of the intestinal immune response, and serve to protect the intestine from pathogenic microorganisms. B lymphocytes mature and differentiate into plasma cells that secrete secretory immunoglobulin A (sIgA) into the intestinal lumen, which is the main immunoglobulin in the intestine and is actively transported to the mucosal surface as a dimer through the polymeric immunoglobulin receptor (pIgR), thereby maintaining the balance and normal function of the intestinal microenvironment [67]. There is a close relationship between the immune system and HAECs. For example, immune organs, immune cells, immunoglobulin, cytokines, inflammasome and exosomes are involved in the occurrence of HaECs (Figure 4).
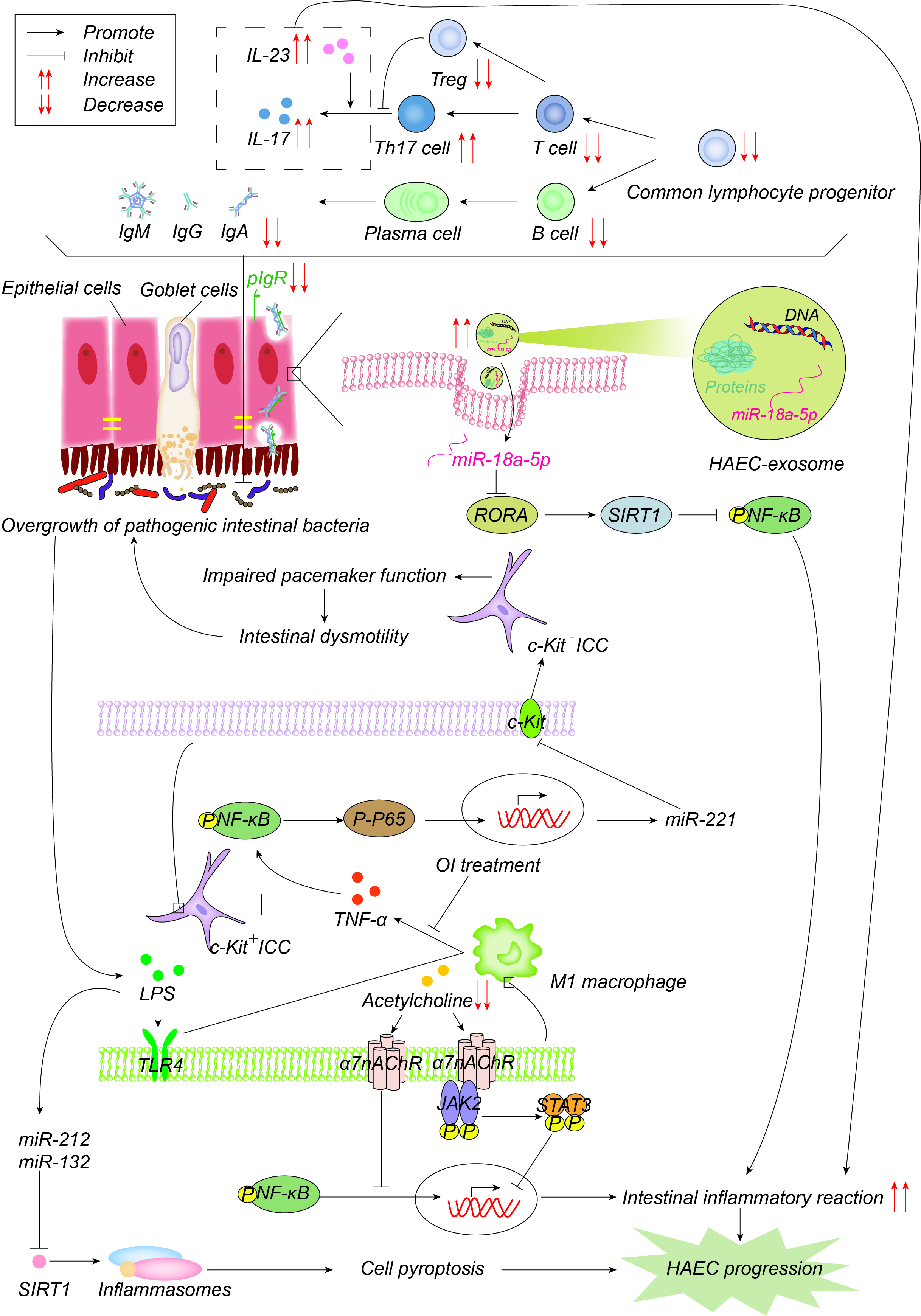
图4. 发病机制在免疫力方面有所提高。常见的淋巴细胞祖细胞群体的数量以及因此T细胞和B细胞的数量显着减少,导致免疫球蛋白减少,其通过聚合的免疫球蛋白受体(pIgR)以二聚体的形式主动转运到粘膜表面,以维持肠道微环境平衡和正常功能。pIgR也会降低,这共同导致肠道微环境紊乱和细菌过度生长。增加的Th17细胞产生更多的IL-17并促进肠道炎症反应。一方面,细菌产生的LPS通过TLR1途径激活M4巨噬细胞,M1巨噬细胞产生TNF-α,通过NF-κB/miR-221途径抑制ICC中的c-Kit表达,导致肠蠕动障碍,进而导致肠道细菌的积累和过度生长。4-辛基衣康酸酯(OI)通过抑制巨噬细胞活化来减少促炎因子的产生并促进ICC表型恢复。另一方面,LPS诱导miR-132/-212的上调,通过抑制sirtuin 3(Sirt3)的表达激活炎性小体NOD-,LRR-和pyrin结构域的含蛋白1(NLRP1),促进细胞焦亡,进而促进HAEC的发生和发展。乙酰胆碱作用于巨噬细胞表面α7烟碱乙酰胆碱受体(α7nAChR),抑制巨噬细胞活化,激活JAK2-STAT3的抗炎途径,抑制NF-κB的炎症途径。HAEC中的乙酰胆碱减少,上述抗炎作用减弱。外泌体miR-18a-5p下调RAR相关孤儿受体A(RORA),激活SIRT1/NF-κB信号通路,诱导过度炎症反应,从而促进HAEC的发展。
This entry is adapted from the peer-reviewed paper 10.3390/ijms24054602
This entry is offline, you can click here to edit this entry!