Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ramendra Pati Pandey | -- | 3020 | 2023-04-24 11:48:29 | | | |
| 2 | Rita Xu | Meta information modification | 3020 | 2023-04-25 04:29:56 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ruby, M.; Gifford, C.C.; Pandey, R.; Raj, V.S.; Sabbisetti, V.S.; Ajay, A.K. Autophagy and Chronic Kidney Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/43382 (accessed on 23 July 2026).
Ruby M, Gifford CC, Pandey R, Raj VS, Sabbisetti VS, Ajay AK. Autophagy and Chronic Kidney Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/43382. Accessed July 23, 2026.
Ruby, Miss, Cody C. Gifford, Ramendrapati Pandey, V. Samuel Raj, Venkata S. Sabbisetti, Amrendra K. Ajay. "Autophagy and Chronic Kidney Disease" Encyclopedia, https://encyclopedia.pub/entry/43382 (accessed July 23, 2026).
Ruby, M., Gifford, C.C., Pandey, R., Raj, V.S., Sabbisetti, V.S., & Ajay, A.K. (2023, April 24). Autophagy and Chronic Kidney Disease. In Encyclopedia. https://encyclopedia.pub/entry/43382
Ruby, Miss, et al. "Autophagy and Chronic Kidney Disease." Encyclopedia. Web. 24 April, 2023.
Copy Citation
Autophagy is a lysosomal protein degradation system that eliminates cytoplasmic components such as protein aggregates, damaged organelles, and even invading pathogens. Autophagy is an evolutionarily conserved homoeostatic strategy for cell survival in stressful conditions and has been linked to a variety of biological processes and disorders. It is vital for the homeostasis and survival of renal cells such as podocytes and tubular epithelial cells, as well as immune cells in the healthy kidney. Autophagy activation protects renal cells under stressed conditions, whereas autophagy deficiency increases the vulnerability of the kidney to injury, resulting in several aberrant processes that ultimately lead to renal failure.
chronic kidney disease
autophagy
TGF-β1
ATG
1. Introduction
Autophagy is a critical homeostatic mechanism that clears a variety of damaged or unwanted intracellular components to protect renal cells under stressed conditions. Autophagy deficiency, on the other hand, increases the vulnerability of the kidney to injury, resulting in reduced renal function, accumulation of damaged mitochondria, severe renal fibrosis, and early kidney failure [1][2]. The role of autophagy in fibrosis has been studied in a variety of ways. A complex communication pathway that detects changes in energy availability for activating or inhibiting autophagy has been identified in several studies [3]. Autophagy and the lysosomal degradation pathway aids in maintaining cellular homeostasis in the kidney cells. Any dysregulation in autophagy causes kidney diseases such as renal fibrosis [4]. Unbalanced autophagy can cause damage to podocytes, proximal tubular cells as well as glomerulosclerosis, as shown in Figure 1. After acute kidney injury, autophagy protects tubular cells from apoptosis and promotes cellular regeneration (as reviewed in [5]), although extended autophagy may lead to excessive digestion of vital cellular components and ultimately cell death [6]. Some of the most well-known proteins in the autophagic membrane are LC3, Beclin1, Atg 7, and Atg 12 [7].
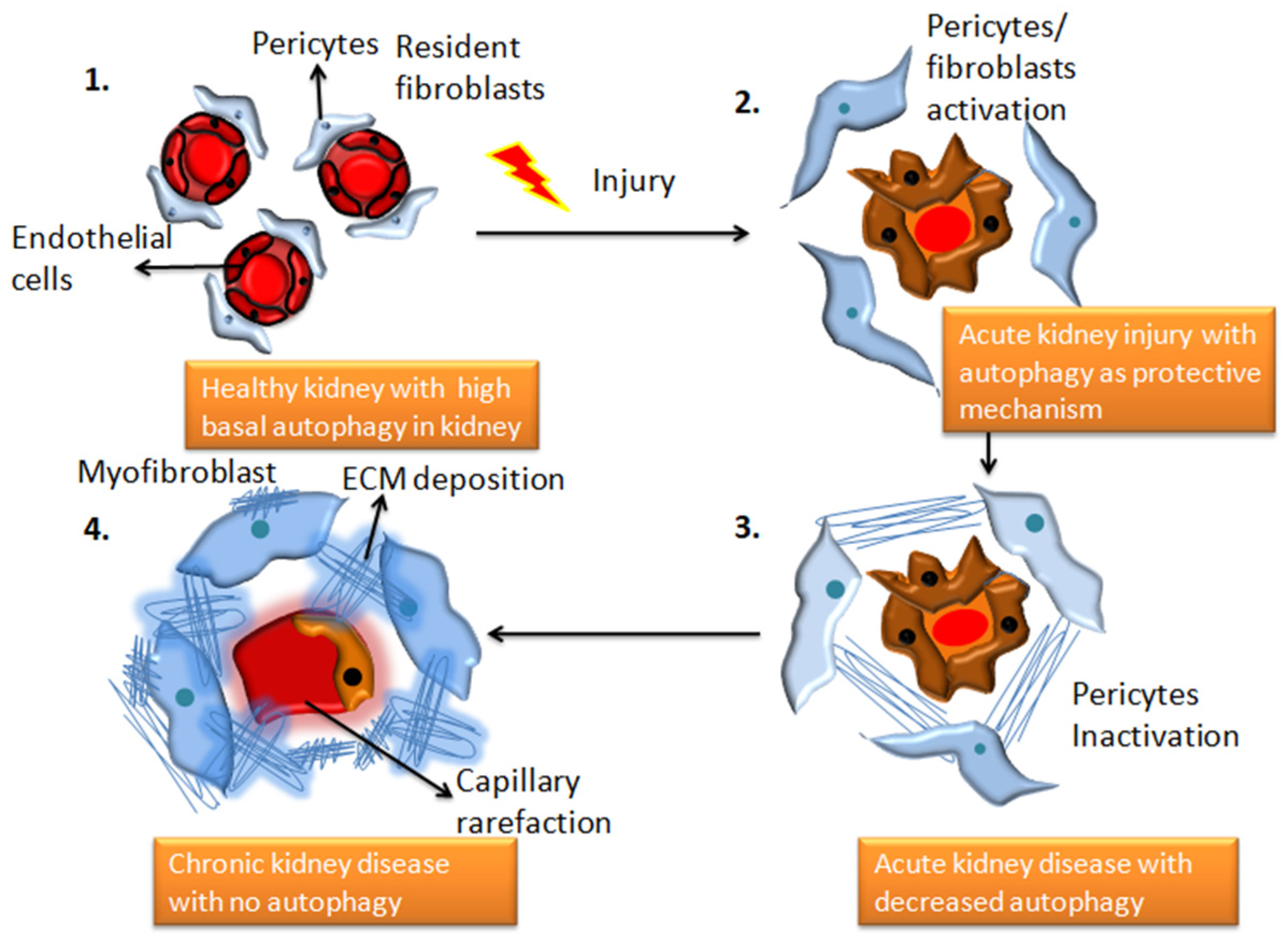
Figure 1. The transition from a healthy kidney to a chronically diseased kidney is associated with changes in autophagy patterns. 1. Demonstrates structurally healthy and physiologically efficient healthy kidney cells with high basal autophagy 2. Depicts the state of renal capillaries following damage caused by an underlying illness, toxins, drugs, or infection. In the extracellular areas, fibroblasts begin to release collagen and fibronectin proteins, and autophagy functions as a protective response, which can be reversible (adaptive repair). 3. AKI worsens to chronic kidney disease, and autophagic activities decline. This stage is characterized by morphological damage to the renal parenchyma and extracellular matrix deposition (maladaptive repair). 4. chronic kidney disease with significant fibrous protein deposition, and no evidence of autophagy.
Kidney fibrosis is identified by uncontrolled production and assembly of extracellular matrix (ECM) proteins in the interstitium of the kidney, causing organizational destruction, functional impairment, and eventually renal failure [8]. The buildup of scar tissue causes the expansion of the cortical-interstitial space, which is a predictor of chronic kidney disease [9]. Although the formation of a fibrotic framework after injury appears to aid tissue regeneration, it is eventually reabsorbed during interstitial remodeling following a mild injury, known as adaptive repair. CKD is characterized by maladaptive epithelial repair, comprising mitochondrial dysfunction, oxidative stress, abnormal autophagy, and tubular growth arrest, and apoptosis at the cellular level. Additionally, fibrotic matrix deposition proceeds unchecked in chronic injuries, disrupting organ design, function, and blood flow. This, in conjunction with nephron loss, causes kidney failure by impairing the body’s capacity to repair the functional tissues (reviewed in [8][9][10]). Numerous stimuli (TGF-β1, WNT, and connective tissue growth factor (CTGF)) and pathogenic factors (injury, diabetes, hypertension) can initiate the process, triggering wound healing and inflammatory signaling cascades that promote interstitial fibrosis [10].
In renal tissues, CKD is linked to significant alterations in cell signaling, such as the activation of TGF-β1, p53, and developmental genes such as Wnt and Notch [7][10]. Regardless of the etiology of kidney fibrogenesis, the TGF-β1/Smad3 signaling pathway is one of the key components that promotes the transcription of several profibrotic genes and drives fibroblast activation [11]. TGF-β activated kinase1 (TAK1), which activates the MAPK kinase (MKK)3-p38, are key upstream signaling molecules in TGF-β1-induced collagen I production [7]. According to a recent study, autophagy modulates the expression of TGF-β1 by proteolytic degradation and suppresses renal tubulointerstitial fibrosis in the unilateral ureteral obstruction (UUO) model. Increased TGF-β1 expression in LC3 deficient and Beclin1 (BECN1) heterozygous mice (autophagy-deficient) correlated with increased collagen I [11]. This study highlights a critical homeostatic negative feedback loop, where TGF-β1 promotes increased fibrogenesis while simultaneously upregulating autophagic processes that degrade mature TGF-β1 to terminate fibrotic signaling events.
WISP-1, a profibrotic protein, may promote renal fibrosis by activating autophagy in both obstructive nephropathy and TGF-β1-treated tubular epithelial cells (TECs) [12]. Autophagy inhibition makes TECs more vulnerable to TGF-β1 induced G1 cell cycle arrest and proliferative reduction, which leads to tubulointerstitial fibrosis (TIF) and nephron loss [13]. Nam, SA et al. found that autophagy in FOXD1-lineage stromal cells protects against renal TIF via downregulating TGF-β1/Smad4 levels through NLRP3 inflammasome signaling [14].
2. Autophagy and Chronic Kidney Disease
2.1. Fibrosis in Chronic Kidney Disease
CKD is the terminal stage of many kidney disorders, characterized by a glomerular filtration rate of less than 60 mL per minute for more than three months, which is driven by chronic changes in kidney morphology and function that has severe consequences for the patient’s health. It contributes to poor medical prognoses and an extreme financial burden on healthcare systems worldwide [15]. Renal fibrosis is the most prevalent clinical outcome of CKD despite the underlying injury or illness, and many signaling networks act simultaneously to influence disease outcomes. Insufficient repair from acute kidney injury has been linked to the worsening of CKD in a growing number of clinical investigations. This is corroborated by the discovery that inadequate tubular regeneration is linked to prolonged tubulointerstitial inflammation, fibroblast activation and proliferation, and extensive extracellular matrix (ECM) protein accumulation. Recent research has also shown that tubule-specific damage is enough to cause fibrosis, making it a key connection between AKI and CKD [16].
Glomerular filtration rate (GFR) is a well-established measure of renal excretory function and albuminuria is a sign of renal barrier failure. GFR and albuminuria are applied to categorize CKD (Glomerular injury). Both have been discovered to be accurate predictive indicators of CKD outcomes in the long run [17]. Numerous studies have linked reduced nephron counts to a higher risk of developing chronic kidney disease [18], and several new hypotheses have been described further to distinguish the relationship between adaptive and maladaptive repair. Tubular cells stalled in a dedifferentiated condition with the continued synthesis of profibrotic factors because of repeated insults following localized tubular epithelial damage contributes to the microvascular loss. JNK activation caused by tubular cells arrested in G2/M leads to fibrotic gene expression [19]. AKI has been linked to changes in DNA methylation and histone modification which results in altered transcription of genes linked to kidney damage, including tumor necrosis factor (TNF). These long-term changes lead to the development of CKD [20]. Kidney injury molecule-1 (KIM-1) is a vital biomarker for renal damage. Tubules positive for KIM-1 correlated with increased macrophage infiltration and pre-fibrotic regions show elevated expression of α-SMA, a fibroblast activation marker [21]. According to Humphreys et al., chronic KIM-1 expression causes inflammation and tubular interstitial fibrosis [22]. Even in normal settings, changes outside the chronically injured kidney lead to a state of relative hypoxia, with fewer peritubular capillaries and higher collagen accumulation, resulting in enhanced gaps between vessels and tubular cells. Reduced number of glomeruli due to injury results in hyperfiltration and higher tubular oxygen uptake in their respective tubules, aggravating the oxygen demand-delivery mismatch [8].
TGF-β1 is a versatile cytokine that has been shown to control a wide range of cellular activities including growth, differentiation, cell death, and healing as well as its important functions in pathology, such as bone disorders, fibrosis, and cancer [23]. TGF-β1 plays a key role in the start and advancement of renal fibrosis; its effector Smad proteins (Smad2, Smad3, and Smad4) have diverse and even antagonistic roles in fibrosis regulation. Smad3 can connect directly to Smad-binding sites within gene promoters to boost transcription; however, neither Smad2 nor Smad4 have DNA binding domains and instead operate as Smad3-based gene transcription regulators [24]. The autophagic factors responsible for inducing and inhibiting kidney fibrosis are depicted in a flow diagram in Figure 2. TGF-β1 signaling has been involved in the pathogenesis of illnesses such as connective tissue disorders, and fibrosis, and it is now well known that TGF-β1 controls a range of essential processes in normal growth and development and physiology [25].
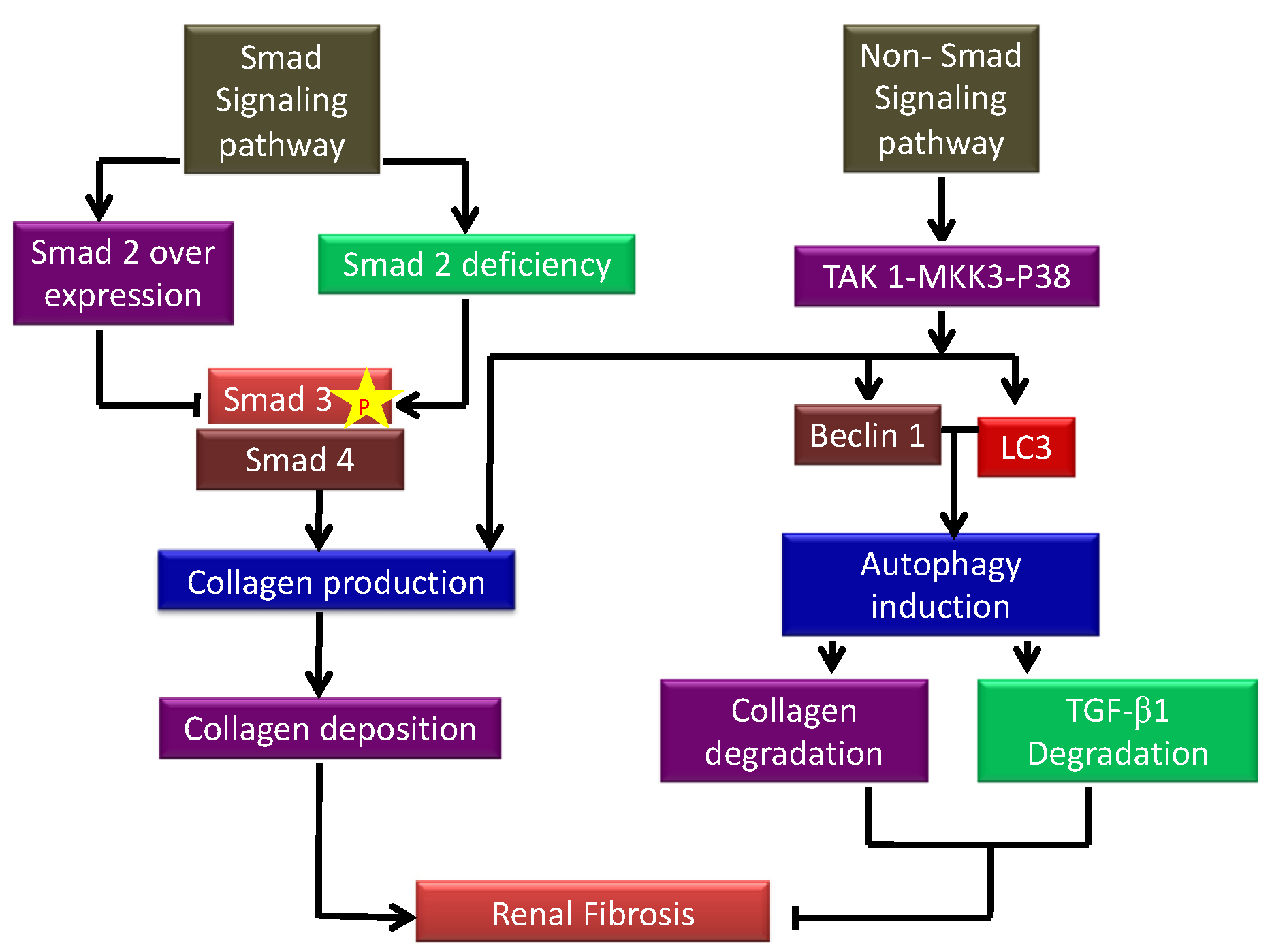
Figure 2. Schematic showing the autophagic factors responsible for inducing and inhibiting kidney fibrosis. p-Smad signaling increases collagen I production deposition leading to the development of renal fibrosis. On the other hand, non-Smad signaling inhibits fibrosis by inhibiting autophagy-mediated degradation of TGF-β1.
Resident fibroblasts produce an interstitial matrix, which helps to stabilize the tubular section of the nephron. A distinct population of myofibroblasts has been regarded as the major origin of ECM in fibrotic development [26]. The functional components of the kidney are maintained by mesenchymal structures. Mesangial cells help to maintain the glomerular tuft of capillaries, which is responsible for the filtration process [27]. Modifications such as podocyte dedifferentiation, podocyte-mediated endothelial dysfunction, and podocyte-induced epithelial-mesenchymal transition all contribute to the development of kidney fibrosis [28]. Furthermore, mesenchymal pericytes aid in vascular repair following damage; however, when parenchymal elements are irrevocably lost throughout the damage process, mesenchymal elements replace the area with an extracellular matrix, ultimately causing kidney fibrosis.
2.2. Autophagy in Chronic Kidney Disease
Podocytes, being terminally differentiated cells, use autophagy rather than cell division to minimize the intracellular buildup of defective DNA and other unwanted macromolecules. Podocytes have a high level of basal autophagy which plays a significant role in maintaining podocyte integrity. Autophagy is a crucial intracellular mechanism for the viability of renal cells. However, either high or defective autophagy may result in podocyte damage. By increasing the activity of systemic NADPH, ANG II has been discovered to increase reactive oxygen species (ROS) generation and oxidative stress in the renal system, resulting in aberrant podocyte autophagy. Autophagy improved survival when cells are treated with ANG II, suggesting that podocytes increasing autophagy might be a potential target treatment for ANG II-induced podocyte damage [29]. The mTOR pathway activates autophagy, which protects phagocytes against apoptosis, foot process effacement, and the development of CKD, and rat and human podocyte studies have suggested that autophagy can be controlled through mTOR signaling [5]. Additionally, endothelial cells have been demonstrated to be regulated by autophagy during the transition process [30].
The loss of autophagy in podocytes significantly increases the sensitivity to several renal injuries. Mice with Atg 5 or Atg 7 loss of function mutations exhibited histological and clinical hallmarks of human focal segmented glomerulosclerosis (FSGS). Podocyte dysfunction was also observed when Atg 5 or Atg 7 is deleted [31]. Proteinuria is associated with a rapid development of end stage renal disease in patients with renal impairments. PTECs are exposed to excess proteins on the luminal surface, which promotes tubular atrophy and fibrosis, contributing to the progression of CKD. Excessive albumin is toxic to PTECs, and defective autophagy contributes to albumin overload-mediated proximal tubular cell toxicity. Albumin exposure, which is common in proteinuric conditions, reduces autophagosome number and impairs autophagosome function through an mTOR-dependent mechanism [32]. Free fatty acids from the bloodstream and the glomerular filtrate are constantly taken up by PTECs and used for ATP generation via beta-oxidation. Regardless, there have not been enough investigations on lipid metabolism in the kidney. A recent discovery demonstrates that autophagy slowdown is a novel mechanism of lipo-toxicity in PTECs [33]. Pharmacological autophagic manipulations are a possible technique for lowering kidney lipo-toxicity. Simple autophagy activation, on one hand, may result in increased lysosomal stress and phospholipid buildup. Autophagy inhibition, on the other hand, can phenocopy autophagy-deficient animals, resulting in increased lipo-toxicity. As a result, the accumulation of pharmaceutical phospholipids that restore autophagic flux might be a unique therapeutic strategy [33].
Autophagy is also involved in the fight against inflammation, which is well-known to be linked to the development of CKD. Autophagy’s anti-inflammatory functions contribute to protection against the development of CKD. Interestingly, autophagy has the ability to modulate a variety of metabolic indices that are linked to renal damage, making it an attractive therapeutic target (reviewed in [34]).
Chronic and progressive kidney failure is caused by fibrosis, and the rate of Incidence is rising globally. Phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) plays a critical role, acting as a signaling pathway to reduce autophagy. The mTOR (molecular receptor of rapamycin), which is highly evolutionarily conserved, is a well-known route that inhibits autophagy. In the obstructed kidneys of rats, autophagy increased followed by interstitial fibrosis, peaking after 3 days of UUO. Suppression of autophagy with 3-MA (3-methyladenine) resulted in reduced Akt/mTOR signaling, exacerbated tubular cell death, and interstitial fibrosis [35]. In other animal models, blunt suppression of mTOR by pharmacological or genetic methods may be advantageous, but due to the interconnectedness of these pathways, such action may have a detrimental impact on autophagy and raise cellular stress in the long run. Autophagy is a cell survival strategy, but it can also contribute to cell death as a result of chronic stress [36]. Autophagy can be profibrotic or antifibrotic depending on the contexts, based on the cell types and pathological circumstances. However, further study is needed to determine the best circumstances for initiating or suppressing autophagy to avoid chronic kidney disease progression.
To evaluate whether targeting autophagy is a realistic technique for the prevention or treatment of kidney illnesses, additional research is needed employing medications that are more selective for autophagy, as well as animal models with inducible autophagy modulation prior, during, and after kidney damage.
2.2.1. TGF-β1 as an Inducer of Autophagy
Recent reports suggest a novel method through which TGF-β1 may provide cytoprotective benefits by inducing macro-autophagy [12][36]. TGF-β1 activated autophagy-related genes Atg5, Atg7, LC3, and Beclin 1 in kidney TECs, resulting in the formation of autophagosomes and the conversion of LC3 to the lipidated form, LC3-II. TGFβ-1 activates autophagy via the TAK1-MKK3-p38 signaling pathway, which increases collagen aggregation and breakdown of insoluble intracellular procollagen, suggesting a role for TGF-β1 in protection from fibrotic kidney disease [37]. Recent reports show that in vivo modulation of Beclin1 in mice protects from AKI-mediated kidney fibrosis development [38]. Livingston et al. reported that FGF2 secretion from injured PTECs PTECs led to the activation of fibroblasts and leads to kidney fibrosis. Fibronectin was deposited in the cells along with autophagy induction [39]. Furthermore, upregulation of WISP-1 in TECs promoted autophagy, as demonstrated by enhanced GFP-LC3 and LC3 and Beclin 1 expression in response to TGF-β1. Reports suggest that TGF-β1 induces autophagy via the TAK1-MKK3-p38 signaling axis, which protects renal mesangial cells from death during serum deprivation [12][40]. Additionally, (TAK1)-binding proteins 2 (TAB2) and TAB3, two of TAK1’s binding partners, have recently been shown to be endogenous autophagy inhibitors [41]. Thus, these reports confirm the role of TGF-β1 as an inducer of autophagy.
2.2.2. Autophagy: A Regulator of TGF-β1
Numerous functions of TGF-β1 demand that its signaling be closely controlled at several levels. TGF-β1 activates Smad7, an inhibitory Smad that regulates TGF-β1 via a negative feedback loop [23]. Autophagy has recently been discovered to have a function in the regulation of the IL-1 family of cytokines. Like TGF-β1, IL-1β is a pro-inflammatory cytokine that is first generated as a pro form and regulates autophagy. IL-1β secretion by directing intracellular pro–IL-1β for destruction. New results point to autophagy as a cytoprotective mechanism that limits TGF-β1 secretion and suppresses the establishment of interstitial fibrosis in kidney injury by negatively regulating the generation of mature TGF-β1 proteins in RPTECs (renal proximal tubular epithelial cells) [23]. Increased TGF-β1 expression was also shown in UUO injured LC3 deficient and BECN1 heterozygous mice, correlated with increased collagen I [11], demonstrating that autophagy can, in fact, regulate expression of TGF-β1, uncovering yet another negative feedback loop, homeostatically programmed to limit TGF-β1 activity during renal injury progression. Thus, it has been shown that TGF-β1 plays a dual role in regulating autophagy, and there is a need to further investigate the role of TGF-β1 in autophagy to understand its true therapeutic potential.
2.2.3. Other Regulators of Autophagy
Several signaling pathways influence autophagy. The mTOR pathway controls macro-autophagy. Ragulator, in conjunction with V-ATPase and Rag, may detect intracellular amino acid levels [42]. The LC3-interacting region (LIR) motif is regulated by post-translational modifications in other autophagy receptors, blocking or increasing the interaction with Atg8 family members. Post-translational modifications influence ATG8 proteins as well [43]. Even though mTORC1 activity is suppressed, autophagy initiation is suppressed, and cap-dependent mRNA translation is sustained during mitosis, according to recent research. A switch from mTORC1 to cyclin-dependent kinase 1 (CDK1)-mediated regulation is a significant contributor to this process [44]. Through its metabolite leucine, acetyl-coenzyme A inhibits autophagosome biogenesis (AcCoA). Acetyl-coenzyme A suppresses autophagy through increasing EP300-dependent acetylation of mTORC1 component raptor, resulting in mTORC1 activation. Interestingly, the major impacts on autophagy in leucine deficiency circumstances are driven by diminished raptor acetylation, which inhibits mTORC1, rather than altered acetylation of other autophagy regulators [45].
A second set of macro-autophagy activation mechanisms has been discovered that is not dependent on mTORC1. One of these mechanisms includes the activation of adenylate cyclase, which increases cAMP levels, allowing inositol 1,4,5-triphosphate (IP3) synthesis via phospholipase C activation. The JNK1/Beclin1/PI3KCIII axis is the third well-known mTOR-independent macro-autophagy regulatory pathway. JNK1-mediated autophagy activation is seen in response to hunger, apoptosis, or elevated nitric oxide levels in the cytosol [46].
2.2.4. Autophagy Deficient Kidney
By digesting and reusing defective macromolecules and organelles, autophagy plays a critical role in metabolic reactions and cellular stability. For example, autophagy defects can promote endothelial-to-mesenchymal transition (EndMT). Autophagy is required for maintaining homeostasis in a variety of organs and cells and is particularly important in the kidney. Conditional autophagy-knockout mice (tissue-specific Atg5- or Atg7-KO mice) and autophagy-deficient cells have solidified the role of basal autophagy in cellular homeostasis in the kidney. In TECs, Atg5 has a detrimental effect on cell cycle progression, and autophagy suppression may enhance renal fibrosis via a G2/M cell cycle arrest-dependent mechanism [47]. In autophagy-deficient proximal tubular cells, protein aggregates and inclusion bodies have been reported [23]. Therefore, PTECs lacking autophagy is more vulnerable to a variety of stresses, and strategies to initiate autophagy when it is absent during fibrosis progression present an effective way to target and limit fibrogenesis and disease development.
References
- Wang, C.-W.; Klionsky, D.J. The Molecular Mechanism of Autophagy. Mol. Med. 2003, 9, 65–76.
- Liang, S.; Wu, Y.S.; Li, D.Y.; Tang, J.X.; Liu, H.F. Autophagy and Renal Fibrosis. Aging Dis. 2022, 13, 712–731.
- Tang, C.; Livingston, M.J.; Liu, Z.; Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 2020, 16, 489–508.
- Zhao, X.C.; Livingston, M.J.; Liang, X.L.; Dong, Z. Cell Apoptosis and Autophagy in Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 557–584.
- Lin, T.A.; Wu, V.C.; Wang, C.Y. Autophagy in Chronic Kidney Diseases. Cells 2019, 8, 61.
- Kaushal, G.P.; Shah, S.V. Autophagy in acute kidney injury. Kidney Int. 2016, 89, 779–791.
- Bao, J.; Shi, Y.; Tao, M.; Liu, N.; Zhuang, S.; Yuan, W. Pharmacological inhibition of autophagy by 3-MA attenuates hyperuricemic nephropathy. Clin. Sci. 2018, 132, 2299–2322.
- Ferenbach, D.A.; Bonventre, J.V. Acute kidney injury and chronic kidney disease: From the laboratory to the clinic. Nephrol. Ther. 2016, 12 (Suppl. 1), S41–S48.
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326.
- He, L.; Wei, Q.; Liu, J.; Yi, M.; Liu, Y.; Liu, H.; Sun, L.; Peng, Y.; Liu, F.; Venkatachalam, M.A.; et al. AKI on CKD: Heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017, 92, 1071–1083.
- Ding, Y.; Kim, S.L.; Lee, S.Y.; Koo, J.K.; Wang, Z.; Choi, M.E. Autophagy regulates TGF-β expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J. Am. Soc. Nephrol. 2014, 25, 2835–2846.
- Yang, X.; Wang, H.; Tu, Y.; Li, Y.; Zou, Y.; Li, G.; Wang, L.; Zhong, X. WNT1-inducible signaling protein-1 mediates TGF-β1-induced renal fibrosis in tubular epithelial cells and unilateral ureteral obstruction mouse models via autophagy. J. Cell Physiol. 2020, 235, 2009–2022.
- Yang, C.; Wu, H.L.; Li, Z.H.; Chen, X.C.; Su, H.Y.; Guo, X.Y.; An, N.; Jing, K.P.; Pan, Q.J.; Liu, H.F. Autophagy Inhibition Sensitizes Renal Tubular Epithelial Cell to G1 Arrest Induced by Transforming Growth Factor beta (TGF-β). Med. Sci. Monit. 2020, 26, e922673.
- Nam, S.A.; Kim, W.Y.; Kim, J.W.; Kang, M.G.; Park, S.H.; Lee, M.S.; Kim, H.W.; Yang, C.W.; Kim, J.; Kim, Y.K. Autophagy in FOXD1 stroma-derived cells regulates renal fibrosis through TGF-β and NLRP3 inflammasome pathway. Biochem. Biophys. Res. Commun. 2019, 508, 965–972.
- Deng, X.; Xie, Y.; Zhang, A. Advance of autophagy in chronic kidney diseases. Ren. Fail. 2017, 39, 306–313.
- Liu, B.C.; Tang, T.T.; Lv, L.L.; Lan, H.Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579.
- Delles, C.; Vanholder, R. Chronic kidney disease. Clin. Sci. 2017, 131, 225–226.
- Wang, X.; Garrett, M.R. Nephron number, hypertension, and CKD: Physiological and genetic insight from humans and animal models. Physiol. Genom. 2017, 49, 180–192.
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543.
- Zager, R.A.; Johnson, A.C. Renal ischemia-reperfusion injury upregulates histone-modifying enzyme systems and alters histone expression at proinflammatory/profibrotic genes. Am. J. Physiol. Renal Physiol. 2009, 296, F1032–F1041.
- van Timmeren, M.M.; van den Heuvel, M.C.; Bailly, V.; Bakker, S.J.; van Goor, H.; Stegeman, C.A. Tubular kidney injury molecule-1 (KIM-1) in human renal disease. J. Pathol. 2007, 212, 209–217.
- Humphreys, B.D.; Xu, F.; Sabbisetti, V.; Grgic, I.; Movahedi Naini, S.; Wang, N.; Chen, G.; Xiao, S.; Patel, D.; Henderson, J.M.; et al. Chronic epithelial kidney injury molecule-1 expression causes murine kidney fibrosis. J. Clin. Investig. 2013, 123, 4023–4035.
- Sureshbabu, A.; Muhsin, S.A.; Choi, M.E. TGF-β signaling in the kidney: Profibrotic and protective effects. Am. J. Physiol. Renal Physiol. 2016, 310, F596–F606.
- Ma, T.T.; Meng, X.M. TGF-β/Smad and Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 347–364.
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873.
- Sun, Y.B.; Qu, X.; Caruana, G.; Li, J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation 2016, 92, 102–107.
- Suárez-Álvarez, B.; Liapis, H.; Anders, H.J. Links between coagulation, inflammation, regeneration, and fibrosis in kidney pathology. Lab. Investig. 2016, 96, 378–390.
- Deelman, L.; Sharma, K. Mechanisms of kidney fibrosis and the role of antifibrotic therapies. Curr. Opin. Nephrol. Hypertens 2009, 18, 85–90.
- Gao, N.; Wang, H.; Yin, H.; Yang, Z. Angiotensin II induces calcium-mediated autophagy in podocytes through enhancing reactive oxygen species levels. Chem. Biol. Interact. 2017, 277, 110–118.
- Hung, T.W.; Chu, C.Y.; Yu, C.L.; Lee, C.C.; Hsu, L.S.; Chen, Y.S.; Hsieh, Y.H.; Tsai, J.P. Endothelial Cell-Specific Molecule 1 Promotes Endothelial to Mesenchymal Transition in Renal Fibrosis. Toxins 2020, 12, 506.
- Oliva Trejo, J.A.; Asanuma, K.; Kim, E.H.; Takagi-Akiba, M.; Nonaka, K.; Hidaka, T.; Komatsu, M.; Tada, N.; Ueno, T.; Tomino, Y. Transient increase in proteinuria poly-ubiquitylated proteins ER stress markers in podocyte-specific autophagy-deficient mice following unilateral nephrectomy. Biochem. Biophys. Res. Commun. 2014, 446, 1190–1196.
- Nolin, A.C.; Mulhern, R.M.; Panchenko, M.V.; Pisarek-Horowitz, A.; Wang, Z.; Shirihai, O.; Borkan, S.C.; Havasi, A. Proteinuria causes dysfunctional autophagy in the proximal tubule. Am. J. Physiol. Renal Physiol. 2016, 311, F1271–F1279.
- Yamamoto, T.; Takabatake, Y.; Minami, S.; Sakai, S.; Fujimura, R.; Takahashi, A.; Namba-Hamano, T.; Matsuda, J.; Kimura, T.; Matsui, I.; et al. Eicosapentaenoic acid attenuates renal lipotoxicity by restoring autophagic flux. Autophagy 2021, 17, 1700–1713.
- Satriano, J.; Sharma, K. Autophagy and metabolic changes in obesity-related chronic kidney disease. Nephrol. Dial. Transplant. 2013, 28 (Suppl. 4), iv29–iv36.
- Kim, W.Y.; Nam, S.A.; Song, H.C.; Ko, J.S.; Park, S.H.; Kim, H.L.; Choi, E.J.; Kim, Y.S.; Kim, J.; Kim, Y.K. The role of autophagy in unilateral ureteral obstruction rat model. Nephrology 2012, 17, 148–159.
- Kaushal, G.P.; Chandrashekar, K.; Juncos, L.A.; Shah, S.V. Autophagy Function and Regulation in Kidney Disease. Biomolecules 2020, 10, 100.
- Kim, S.I.; Na, H.J.; Ding, Y.; Wang, Z.; Lee, S.J.; Choi, M.E. Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-β1. J. Biol. Chem. 2012, 287, 11677–11688.
- Shi, M.; Maique, J.; Shepard, S.; Li, P.; Seli, O.; Moe, O.W.; Hu, M.C. In vivo evidence for therapeutic applications of beclin 1 to promote recovery and inhibit fibrosis after acute kidney injury. Kidney Int. 2022, 101, 63–78.
- Livingston, M.J.; Ding, H.F.; Huang, S.; Hill, J.A.; Yin, X.M.; Dong, Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 2016, 12, 976–998.
- Ding, Y.; Kim, J.K.; Kim, S.I.; Na, H.J.; Jun, S.Y.; Lee, S.J.; Choi, M.E. TGF-1 protects against mesangial cell apoptosis via induction of autophagy. J. Bio. Chem. 2010, 285, 37909–37919.
- Takaesu, G.; Kobayashi, T.; Yoshimura, A. TGFβ-activated kinase 1 (TAK1)-binding proteins (TAB) 2 and 3 negatively regulate autophagy. J. Biochem. 2012, 151, 157–166.
- Andrzejewska, Z.; Nevo, N.; Thomas, L.; Chhuon, C.; Bailleux, A.; Chauvet, V.; Courtoy, P.J.; Chol, M.; Guerrera, I.C.; Antignac, C. Cystinosin is a Component of the Vacuolar H+-ATPase-Ragulator-Rag Complex Controlling Mammalian Target of Rapamycin Complex 1 Signaling. J. Am. Soc. Nephrol. 2016, 27, 1678–1688.
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays Biochem. 2017, 61, 609–624.
- Odle, R.I.; Florey, O.; Ktistakis, N.T.; Cook, S.J. CDK1, the Other ‘Master Regulator’ of Autophagy. Trends Cell Biol. 2021, 31, 95–107.
- Son, S.M.; Park, S.J.; Stamatakou, E.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Leucine regulates autophagy via acetylation of the mTORC1 component raptor. Nat. Commun. 2020, 11, 3148.
- Gros, F.; Muller, S. Pharmacological regulators of autophagy and their link with modulators of lupus disease. Br. J. Pharmacol. 2014, 171, 4337–4359.
- Bonventre, J.V. Maladaptive proximal tubule repair: Cell cycle arrest. Nephron. Clin. Pract. 2014, 127, 61–64.
More
Information
Subjects:
Biotechnology & Applied Microbiology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.8K
Revisions:
2 times
(View History)
Update Date:
25 Apr 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No