Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ramendra Pati Pandey and Version 2 by Rita Xu.
Autophagy is a lysosomal protein degradation system that eliminates cytoplasmic components such as protein aggregates, damaged organelles, and even invading pathogens. Autophagy is an evolutionarily conserved homoeostatic strategy for cell survival in stressful conditions and has been linked to a variety of biological processes and disorders. It is vital for the homeostasis and survival of renal cells such as podocytes and tubular epithelial cells, as well as immune cells in the healthy kidney. Autophagy activation protects renal cells under stressed conditions, whereas autophagy deficiency increases the vulnerability of the kidney to injury, resulting in several aberrant processes that ultimately lead to renal failure.
- chronic kidney disease
- autophagy
- TGF-β1
- ATG
1. Introduction
Autophagy is a critical homeostatic mechanism that clears a variety of damaged or unwanted intracellular components to protect renal cells under stressed conditions. Autophagy deficiency, on the other hand, increases the vulnerability of the kidney to injury, resulting in reduced renal function, accumulation of damaged mitochondria, severe renal fibrosis, and early kidney failure [1][2][1,2]. The role of autophagy in fibrosis has been studied in a variety of ways. A complex communication pathway that detects changes in energy availability for activating or inhibiting autophagy has been identified in several studies [3]. Autophagy and the lysosomal degradation pathway aids in maintaining cellular homeostasis in the kidney cells. Any dysregulation in autophagy causes kidney diseases such as renal fibrosis [4]. Unbalanced autophagy can cause damage to podocytes, proximal tubular cells as well as glomerulosclerosis, as shown in Figure 1. After acute kidney injury, autophagy protects tubular cells from apoptosis and promotes cellular regeneration (as reviewed in [5]), although extended autophagy may lead to excessive digestion of vital cellular components and ultimately cell death [6]. Some of the most well-known proteins in the autophagic membrane are LC3, Beclin1, Atg 7, and Atg 12 [7].
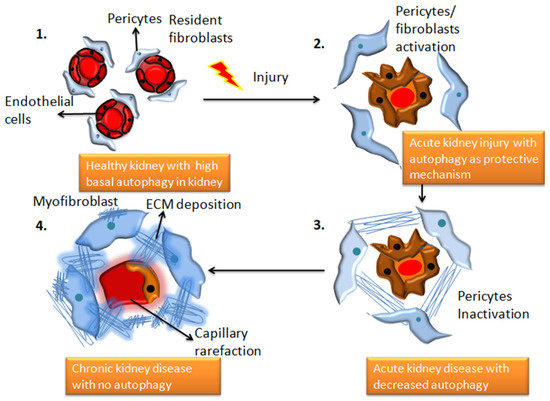
Figure 1. The transition from a healthy kidney to a chronically diseased kidney is associated with changes in autophagy patterns. 1. Demonstrates structurally healthy and physiologically efficient healthy kidney cells with high basal autophagy 2. Depicts the state of renal capillaries following damage caused by an underlying illness, toxins, drugs, or infection. In the extracellular areas, fibroblasts begin to release collagen and fibronectin proteins, and autophagy functions as a protective response, which can be reversible (adaptive repair). 3. AKI worsens to chronic kidney disease, and autophagic activities decline. This stage is characterized by morphological damage to the renal parenchyma and extracellular matrix deposition (maladaptive repair). 4. chronic kidney disease with significant fibrous protein deposition, and no evidence of autophagy.
Kidney fibrosis is identified by uncontrolled production and assembly of extracellular matrix (ECM) proteins in the interstitium of the kidney, causing organizational destruction, functional impairment, and eventually renal failure [8]. The buildup of scar tissue causes the expansion of the cortical-interstitial space, which is a predictor of chronic kidney disease [9]. Although the formation of a fibrotic framework after injury appears to aid tissue regeneration, it is eventually reabsorbed during interstitial remodeling following a mild injury, known as adaptive repair. CKD is characterized by maladaptive epithelial repair, comprising mitochondrial dysfunction, oxidative stress, abnormal autophagy, and tubular growth arrest, and apoptosis at the cellular level. Additionally, fibrotic matrix deposition proceeds unchecked in chronic injuries, disrupting organ design, function, and blood flow. This, in conjunction with nephron loss, causes kidney failure by impairing the body’s capacity to repair the functional tissues (reviewed in [8][9][10][8,9,10]). Numerous stimuli (TGF-β1, WNT, and connective tissue growth factor (CTGF)) and pathogenic factors (injury, diabetes, hypertension) can initiate the process, triggering wound healing and inflammatory signaling cascades that promote interstitial fibrosis [10].
In renal tissues, CKD is linked to significant alterations in cell signaling, such as the activation of TGF-β1, p53, and developmental genes such as Wnt and Notch [7][10][7,10]. Regardless of the etiology of kidney fibrogenesis, the TGF-β1/Smad3 signaling pathway is one of the key components that promotes the transcription of several profibrotic genes and drives fibroblast activation [11]. TGF-β activated kinase1 (TAK1), which activates the MAPK kinase (MKK)3-p38, are key upstream signaling molecules in TGF-β1-induced collagen I production [7]. According to a recent study, autophagy modulates the expression of TGF-β1 by proteolytic degradation and suppresses renal tubulointerstitial fibrosis in the unilateral ureteral obstruction (UUO) model. Increased TGF-β1 expression in LC3 deficient and Beclin1 (BECN1) heterozygous mice (autophagy-deficient) correlated with increased collagen I [11]. This study highlights a critical homeostatic negative feedback loop, where TGF-β1 promotes increased fibrogenesis while simultaneously upregulating autophagic processes that degrade mature TGF-β1 to terminate fibrotic signaling events.
WISP-1, a profibrotic protein, may promote renal fibrosis by activating autophagy in both obstructive nephropathy and TGF-β1-treated tubular epithelial cells (TECs) [12]. Autophagy inhibition makes TECs more vulnerable to TGF-β1 induced G1 cell cycle arrest and proliferative reduction, which leads to tubulointerstitial fibrosis (TIF) and nephron loss [13]. Nam, SA et al. found that autophagy in FOXD1-lineage stromal cells protects against renal TIF via downregulating TGF-β1/Smad4 levels through NLRP3 inflammasome signaling [14].
2. Autophagy and Chronic Kidney Disease
2.1. Fibrosis in Chronic Kidney Disease
CKD is the terminal stage of many kidney disorders, characterized by a glomerular filtration rate of less than 60 mL per minute for more than three months, which is driven by chronic changes in kidney morphology and function that has severe consequences for the patient’s health. It contributes to poor medical prognoses and an extreme financial burden on healthcare systems worldwide [15][46]. Renal fibrosis is the most prevalent clinical outcome of CKD despite the underlying injury or illness, and many signaling networks act simultaneously to influence disease outcomes. Insufficient repair from acute kidney injury has been linked to the worsening of CKD in a growing number of clinical investigations. This is corroborated by the discovery that inadequate tubular regeneration is linked to prolonged tubulointerstitial inflammation, fibroblast activation and proliferation, and extensive extracellular matrix (ECM) protein accumulation. Recent research has also shown that tubule-specific damage is enough to cause fibrosis, making it a key connection between AKI and CKD [16][47]. Glomerular filtration rate (GFR) is a well-established measure of renal excretory function and albuminuria is a sign of renal barrier failure. GFR and albuminuria are applied to categorize CKD (Glomerular injury). Both have been discovered to be accurate predictive indicators of CKD outcomes in the long run [17][48]. Numerous studies have linked reduced nephron counts to a higher risk of developing chronic kidney disease [18][49], and several new hypotheses have been described further to distinguish the relationship between adaptive and maladaptive repair. Tubular cells stalled in a dedifferentiated condition with the continued synthesis of profibrotic factors because of repeated insults following localized tubular epithelial damage contributes to the microvascular loss. JNK activation caused by tubular cells arrested in G2/M leads to fibrotic gene expression [19][50]. AKI has been linked to changes in DNA methylation and histone modification which results in altered transcription of genes linked to kidney damage, including tumor necrosis factor (TNF). These long-term changes lead to the development of CKD [20][51]. Kidney injury molecule-1 (KIM-1) is a vital biomarker for renal damage. Tubules positive for KIM-1 correlated with increased macrophage infiltration and pre-fibrotic regions show elevated expression of α-SMA, a fibroblast activation marker [21][52]. According to Humphreys et al., chronic KIM-1 expression causes inflammation and tubular interstitial fibrosis [22][53]. Even in normal settings, changes outside the chronically injured kidney lead to a state of relative hypoxia, with fewer peritubular capillaries and higher collagen accumulation, resulting in enhanced gaps between vessels and tubular cells. Reduced number of glomeruli due to injury results in hyperfiltration and higher tubular oxygen uptake in their respective tubules, aggravating the oxygen demand-delivery mismatch [8]. TGF-β1 is a versatile cytokine that has been shown to control a wide range of cellular activities including growth, differentiation, cell death, and healing as well as its important functions in pathology, such as bone disorders, fibrosis, and cancer [23][54]. TGF-β1 plays a key role in the start and advancement of renal fibrosis; its effector Smad proteins (Smad2, Smad3, and Smad4) have diverse and even antagonistic roles in fibrosis regulation. Smad3 can connect directly to Smad-binding sites within gene promoters to boost transcription; however, neither Smad2 nor Smad4 have DNA binding domains and instead operate as Smad3-based gene transcription regulators [24][55]. The autophagic factors responsible for inducing and inhibiting kidney fibrosis are depicted in a flow diagram in Figure 2. TGF-β1 signaling has been involved in the pathogenesis of illnesses such as connective tissue disorders, and fibrosis, and it is now well known that TGF-β1 controls a range of essential processes in normal growth and development and physiology [25][56].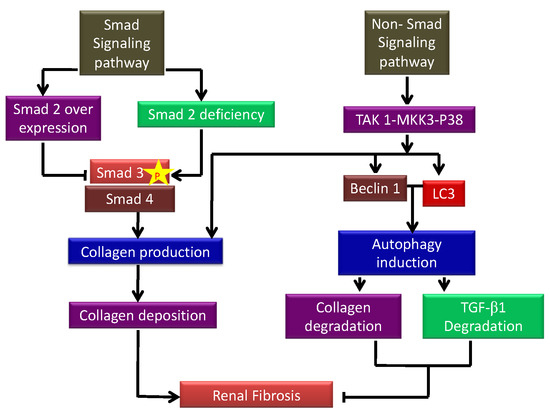
Figure 2. Schematic showing the autophagic factors responsible for inducing and inhibiting kidney fibrosis. p-Smad signaling increases collagen I production deposition leading to the development of renal fibrosis. On the other hand, non-Smad signaling inhibits fibrosis by inhibiting autophagy-mediated degradation of TGF-β1.