Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mahmoud Kandeel | -- | 2606 | 2023-04-18 22:42:52 | | | |
| 2 | Sirius Huang | Meta information modification | 2606 | 2023-04-19 03:00:51 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kandeel, M. miRNA-Based Therapy for Oncogenic Viruses. Encyclopedia. Available online: https://encyclopedia.pub/entry/43214 (accessed on 24 June 2026).
Kandeel M. miRNA-Based Therapy for Oncogenic Viruses. Encyclopedia. Available at: https://encyclopedia.pub/entry/43214. Accessed June 24, 2026.
Kandeel, Mahmoud. "miRNA-Based Therapy for Oncogenic Viruses" Encyclopedia, https://encyclopedia.pub/entry/43214 (accessed June 24, 2026).
Kandeel, M. (2023, April 18). miRNA-Based Therapy for Oncogenic Viruses. In Encyclopedia. https://encyclopedia.pub/entry/43214
Kandeel, Mahmoud. "miRNA-Based Therapy for Oncogenic Viruses." Encyclopedia. Web. 18 April, 2023.
Copy Citation
Approximately 20% of all cases of human cancer are caused by viral infections. Although a great number of viruses are capable of causing a wide range of tumors in animals, only seven of these viruses have been linked to human malignancies and are presently classified as oncogenic viruses. These include the Epstein–Barr virus (EBV), human papillomavirus (HPV), hepatitis B virus (HBV), hepatitis C virus (HCV), Merkel cell polyomavirus (MCPyV), human herpesvirus 8 (HHV8), and human T-cell lymphotropic virus type 1 (HTLV-1). It is possible that virally encoded microRNAs (miRNAs), which are ideal non-immunogenic tools for viruses, play a significant role in carcinogenic processes.
oncogenic viruses
viral miRNAs
cancer
human neoplasms
1. Oncogenic Viral Infections
There are different types of DNA and RNA viruses that are categorized as oncogenic. They cause cancer by a number of methods, one of which is the encoding of miRNAs (Figure 1). Several of the viruses known to be involved in cancer induction or progression are described below.
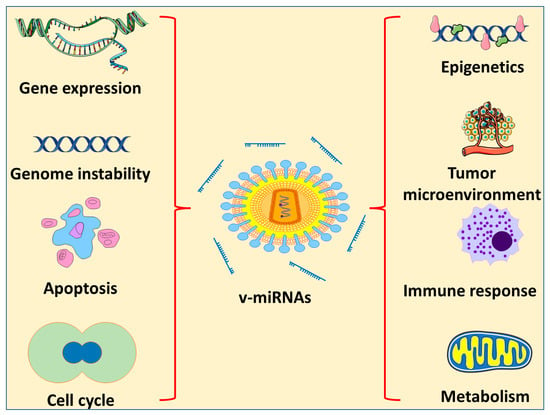
Figure 1. Effects of v-miRNA on different characteristics of cancer progression.
The herpesviruses family includes EBV. This virus is an enveloped virus that, similar to other herpesviruses, has a DNA core surrounded by an icosahedral nucleocapsid and an associated tegument. It is now common knowledge that over 90% of the adult population around the world have contracted EBV at some point, and once they have been infected, they continue to harbor the virus throughout their lives [1]. An exchange of saliva is the mode of transmission for EBV. This oncogenic virus does most of its infecting and replicating in the stratified squamous epithelium of the oropharynx during an acute infection [2][3]. After this, the B cells become infected with a dormant form of the virus (although the sequence of epithelial versus lymphoid infection is a matter of debate). It is believed that the infection of B lymphocytes by EBV takes place in the lymphoid organs of the oropharynx, and in normal carriers, the virus is assumed to survive in circulating memory B cells [4]. Research on epidemiology and immunology suggests that EBV infection may have a role in the development of endemic Burkitt lymphoma. The epithelial cancer known as nasopharyngeal carcinoma is connected with EBV and is prevalent in South China and Southeast Asia [5]. More than 97% of patients with nasopharyngeal carcinoma have a positive EBV test, and a close connection between nasopharyngeal carcinoma and EBV is observed all over the world [6]. In addition to infection, EBV can cause a prevalent kind of cancer called EBV-associated gastric cancer [7], which has a regional distribution of 1.3% to 30.9% of all cases of gastric cancer and a global average of 8.9% of all cases of gastric cancer, or approximately 75,000 new cases each year [8]. Because EBV is associated with the production of certain subsets of latent proteins, the establishment of a latent infection by EBV has been linked to a number of different types of cancer. Epstein–Barr nuclear antigens (EBNAs) 1, 2, 3A, 3B, 3C, and LP are expressed during latent infection in host cells [9]. EBNAs are multi-functional proteins and considered as the only proteins expressed in all types of EBV and found in EBV-associated malignancies [10]. The positive and negative regulation of viral promoters by EBNA1 is essential for gene control, extrachromosomal replication, and maintenance of the EBV episomal genome [11].
The human cytomegalovirus (HCMV) is a member of the Herpesviridae family and has a DNA genome that is 236 kilobase pairs in size [12]. HCMV gene products interfere with cell cycle progression, promote mutation and genomic instability in the viral genome, enhance cell survival, and facilitate immune evasion and tumor growth [13]. In addition, HCMV can infect a wide variety of cell types found in tumors and their microenvironments. HCMV-induced oncomodulation was predicted after the discovery of viral proteins and DNA in several cancer tissues [14]. HepG2 cells that had been infected with HCMV, for instance, were observed to secrete IL-6 in conjunction with autocrine and/or paracrine activation of the IL–6R–JAK–STAT3 pathway. In HepG2 cells that had been infected with HCMV, an increase in cell proliferation concurrently occurred with an increase in the synthesis of cyclin D1 and survivin. Compared with cultures that were not infected with HCMV, cultures that were infected with HCMV had an increase in the development of tumorspheres in HepG2 cells [15]. In addition, when the herpes simplex virus type 1 (HCMV) infected the “stem-like” colorectal cancer HT29 and SW480 cells, both the epithelial–mesenchymal transmission (EMT) pathways and the WNT pathways were activated, which led to increased cellular proliferation and motility [16].
The Papillomaviridae family includes HPV. The DNA genome is double-stranded and contained in non-enveloped virions that make up the virus. An icosahedral capsid that is comprised of the major and minor structural proteins L1 and L2, respectively, encloses the genetic material. These viruses have a very precise tissue preference and can infect the epithelium of the skin as well as the mucosa. On the basis of the genomic sequence of the L1 gene, which codes for the primary capsid protein, over 200 different forms of HPV have been discovered and characterized [17]. Of these, at least 14 high-risk types have the potential to cause cancer. Two kinds of HPVs are responsible for the majority of HPV-related malignancies (productive for CIN1 or abortive for CIN3), including about 70% of cervical cancers and precancerous cervical lesions [18]. Integration often ends in the deregulation of the expression of the viral E6 and E7 oncogenes, which in turn stimulates cellular proliferation, eliminates cell cycle checkpoints, and ultimately leads to increasing genetic instability. This provides cells with an advantage in selective proliferation and increases the propagation of oncogenic mutations [19]. The significance of abnormally dysregulated oncogene expression is shown by the clonal expansion of cells that have incorporated HPV. In fact, the continuing growth and survival of HPV-associated malignancies is contingent on the viral E6 and E7 oncogenes being expressed [20].
HBV, which belongs to the family Hepadnaviridae, has a genome that is 3.2 kilobase pairs in size and contains partly double-stranded DNA. There are four key open reading frames (ORFs) in the HBV genome. These ORFs encode for the polymerase; the surface protein HBsAg, a core protein that forms the nucleocapsid; and the HBV X protein, which is critical in viral replication [21][22]. In eastern Asia and sub-Saharan Africa, infection with endemic HBV is the primary cause of HCC, accounting for around 70% of all cases.
The prevalence of HCV infection in the nations of Europe and North America varies from 50% to 70%, whereas alcohol abuse, which can lead to alcoholic steatohepatitis (ASH), is responsible for around 20% of all cases [23]. The development of HCC typically occurs as a result of a protracted and chronic disease course that is accompanied by underlying liver cirrhosis (~80%) [24]. However, approximately 20% of HCC cases develop in livers that do not have cirrhosis. Only sexual contact with blood or other bodily fluids, or vertical transmission from mother to child, is capable of transmitting HBV, and only a small number of HBV virions are needed to initiate an infection. Most chronic carriers acquire their status because of infection during pregnancy, delivery, or early childhood, when the immune system is still developing. Between 1% and 5% of adults and adolescents who contract an infection will develop a chronic condition [25].
The polyprotein encoded by the HCV is translated and then cleaved into structural (S) and non-structural (NS) proteins. The HCV is a single-stranded, positive-sense RNA virus. Some examples of structural proteins are the core protein, the envelope E1 and E2 glycoproteins, and the p7 protein. Non-structural proteins (NS1, NS2, NS3, NS4A/B, and NS5A/B) aid in viral genome replication and particle assembly. Because it does not encode oncoproteins and integrate its genome into the chromosomal DNA of the host, HCV is unique among cancer-causing viruses. The hepatocyte cytoplasm is the replication site for HCV [26]. It was previously believed that HCV-related HCC development occurred primarily through indirect mechanisms, such as the effects of chronic inflammation and oxidative stress, because HCV RNA cannot integrate into the human genome. Eventually, this condition leads to fibrosis and cirrhosis, as is the case with other causes of HCC such as ASH, non-alcoholic steatohepatitis (NASH), and obesity-related illnesses. The viral proteins themselves, however, have been demonstrated to have a direct oncogenic impact, according to recent studies [27].
The hepatitis delta virus (HDV) is a small circular single-stranded RNA satellite virus that is dependent on HBV for progeny virus production. The HBV envelope glycoprotein is required for HDV particle assembly. As a result, HDV can only produce an infection if there is also an HBV infection present. The most severe form of viral hepatitis is caused by HDV, which affects between 15 and 20 million people throughout the world [28].
It has been proven that immunodeficiency is a risk factor for the development of cancer, and it is probable that the underlying causes are numerous. Some of these possible explanations include the uncontrolled growth of oncogenic viruses and insufficient immune surveillance. The presence of CD4 T cells, as well as their quantity and functionality, are critical components in multiple stages of the oncogenic pathway [29]. These stages include the recognition of tumor antigens, the production of an antibody capable of effectively neutralizing the tumor, cellular responses to viral pathogens, and the elimination of premalignant lesions. Human immunodeficiency virus (HIV-1) carriers have an increased risk of developing a number of cancers, including non-Hodgkin lymphoma, Kaposi sarcoma, cervical cancer, and other cancers connected to chronic viral infections. This has historically been linked to immunological suppression brought on by HIV-1, which is characterized by a decline in CD4+ T-helper cells, exhaustion of lymphopoiesis, and defective lymphocytes. Antiretroviral treatment initiated early could not prevent the development of oncologic problems in the long term. This showed that HIV-1 and its antigens are actively involved in carcinogenesis and may have an influence even at extremely low levels on the background of a rebuilt immune system [30].
The Kaposi sarcoma tissue that was initially examined for the presence of KSHV or HHV-8 was obtained from individuals who suffered from AIDS. There is a correlation between having an infection with KSHV and having certain inflammatory diseases. KSHV has been found to be present, in addition to KS, in primary effusion lymphoma, KSHV-associated lymphoma, and some instances of multicentric Castleman disease [31]. The majority of original KSHV infections do not produce any clinical signs, and similar to other cancers resulting from human oncogenic viral infections, Kaposi sarcoma does not manifest itself until decades after the virus has been dormant. In addition, asymptomatic oral shedding and transmission through body fluids are both possible modes of KSHV transmission [32].
MCV is thought to be the cause of Merkel cell carcinoma, which is an uncommon but aggressive form of skin cancer [33]. The sequencing of the virus recovered from Merkel cell tumors contains tumor-specific mutations that terminate the MCV T antigen. These mutations (which are not present in the wild-type virus recovered from non-tumor sites) inactivate the T antigen helicase, leaving the integrated virus incapable of replicating outside of the host cancer cell. As a result, the tumor acts as a “dead-end host” for MCV [34].
HTLV-1 is a retrovirus associated with several diseases, including lymphomas and myelopathies. It is thought that 1% to 5% of HTLV-1-infected cases develop cancers [35], such as. adult T-cell lymphoma/leukemia (ATLL) and cutaneous T-cell lymphoma.
2. miRNA-Based Therapy for Oncogenic Viruses
The use of miRNAs and anti-miRNA oligonucleotides (AMOs) as potential new treatments has been proposed [36]. miRNAs account for only approximately 3% of human genes, yet they may influence up to 30% of human genes that code for proteins. One intriguing therapeutic idea is that a single miRNA can negatively affect several target proteins by interacting with various target mRNAs [37]. Normalizing the aberrant levels of miRNAs could result in the recovery of affected cells, prevent tumor differentiation, and prohibit cancer metastasis [37]. In this context, miRNA expression alterations could be dealt with in the following two primary ways: (1) degradation of the overexpressed mRNA via miRNA, and (2) regulation of miRNA expression. Injecting miRNAs to make up for the inadequate production of miRNAs is a feasible option.
Yang et al. (2013) devised a plan for combatting HCV infection that involves inserting sequences from the HCV genome into five endogenous miRNAs from a naturally occurring miRNA cluster (miR-17-92). This miRNA cluster (HCV-miR-Cluster 5) was supplied to cells using adeno-associated virus (AAV) vectors, and it made sense that it would be assembled in the liver, where HCV replication and assembly take place. AAV-HCV-miR-Cluster 5 was able to reduce HCV multiplication by 95% in 2 days in vitro, and the steady production of anti-HCV miRNAs blocked HCV from spreading to uninfected cells [38].
2.1. Anti-miR
With respect to tumor progression, miRNA can be viewed as having two classes, tumor-suppressor miRNA and tumor-inducer miRNA. The overexpression of tumor-enhancing miRNA can be overcome by the application of anti-miR. This can be accomplished by the administration of the artificial antisense oligonucleotide. One example of an upregulated miRNA in an oncogenic viral infection is miR149 in HCV infection [39]. The first anti-miR compound introduced to clinical trials was miravirsen, which was used to treat HCV by targeting the liver miR122 [40]. MiRNA sponges are a novel class of miRNA inhibitors, comprising transcripts with tandem repeats of specific sequences for binding a desired miRNA [41]. They act by sequestering the overexpressed miRNA.
EBV BART miRNAs are important targets for treating EBV-associated epithelial tumors because they are overexpressed in these cases [42]. Gold nanoparticles bearing anti-EBV-miR-BART7-3p were successful in decreasing the growth of cancer cells [43].
Anti-miRs are useful because they can be directed against specific miRNAs that play a role in controlling EBV gene expression, replication, and latency. There are several examples of viral miRNAs that have been proven to promote EBV replication and carcinogenesis, including EBV miR-BART6-3p and EBV miR-BART16-5p, miR-BHRF1-2-5p, miR-BART1, BART2, and BART22. As a potential treatment for EBV-associated malignancies, anti-miRs targeting these miRNAs can be created and evaluated in preclinical trials.
Some studies have reported the upregulation of certain miRNAs in HPV-associated cancers. For example, in cervical cancer associated with high-risk HPV types such as HPV-16 and HPV-18, miR-9, miR-21, miR-27b and miR-34a [44] have been reported to be upregulated.
Studies have shown that some of the HHV-8-encoded miRNAs were upregulated in cancer cells, suggesting that they may have contributed to the development and progression of these cancers. For example, the HHV-8 miRNA known as miR-K12-11 has been shown to promote the growth and survival of cancer cells by targeting tumor suppressor genes [45]. A specific anti-miR specific for miR-K12-11 can be a promising target against oncogenic herpesviruses.
2.2. miR Mimetics
miR mimetics are synthetic molecules designed to mimic the function of endogenous miRNAs. These molecules can be used to regulate the expression of specific genes and have potential applications in the treatment of various diseases. The lowered expression of certain miRNAs can enhance the tumorigenic environment. In such therapies, miR mimetics are usually developed for the downregulated host miRNAs. In this case, treatment occurs by the compensation of lowered levels of these miRNAs [41]. Examples of downregulated miRNAs in oncogenic HCV infections include miR29a, miR29b, miR29c, miR17, miR106a, miR106b, miR181a, miR93, miR221, and miR222 [46]. In another example, HCV replication was adversely affected by the overexpression of miR-199a in its binding to the viral 5′UTR [47].
miR-28-3p was shown to target a site inside the HTLV-1 viral gag/pol mRNA, where it suppressed viral growth and mRNA translation and caused infected cells to be less likely to replicate. An abortive infection was caused by miR-28-3p expression, which inhibited HTLV-1 reverse transcription and prevented the formation of the preintegration complex [48].
The viral oncoprotein E6 of oncogenic HPV inhibited the expression of tumor-suppressing miR-34a [49]. Using miR-34a mimetics is expected to be effective in treating HPV-associated cancers.
References
- Day, N.; Geser, A.; Lavoué, M.; Ho, J.; Simons, M.; Sohier, R.; Tukei, P.; Vonka, V.; Zavadova, H. Sero-epidemiology of the Epstein-Barr virus: Preliminary analysis of an international study—A review. IARC Sci. Publ. 1975, 11, 3–16.
- Murray, P.; Young, L. The role of the Epstein-Barr virus in human disease. Front. Biosci. Landmark 2002, 7, 519–540.
- Sixbey, J.W.; Nedrud, J.G.; Raab-Traub, N.; Hanes, R.A.; Pagano, J.S. Epstein–Barr virus replication in oropharyngeal epithelial cells. N. Engl. J. Med. 1984, 310, 1225–1230.
- Thompson, M.P.; Kurzrock, R. Epstein-Barr virus and cancer. Clin. Cancer Res. 2004, 10, 803–821.
- Mahdavifar, N.; Ghoncheh, M.; Mohammadian-Hafshejani, A.; Khosravi, B.; Salehiniya, H. Epidemiology and inequality in the incidence and mortality of nasopharynx cancer in Asia. Osong Public Health Res. Perspect. 2016, 7, 360–372.
- Chang, C.M.; Kelly, J.Y.; Mbulaiteye, S.M.; Hildesheim, A.; Bhatia, K. The extent of genetic diversity of Epstein-Barr virus and its geographic and disease patterns: A need for reappraisal. Virus Res. 2009, 143, 209–221.
- Young, L.S.; Rickinson, A.B. Epstein–Barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768.
- Yang, J.; Liu, Z.; Zeng, B.; Hu, G.; Gan, R. Epstein–Barr virus-associated gastric cancer: A distinct subtype. Cancer Lett. 2020, 495, 191–199.
- AlQarni, S.; Al-Sheikh, Y.; Campbell, D.; Drotar, M.; Hannigan, A.; Boyle, S.; Herzyk, P.; Kossenkov, A.; Armfield, K.; Jamieson, L. Lymphomas driven by Epstein–Barr virus nuclear antigen-1 (EBNA1) are dependant upon Mdm2. Oncogene 2018, 37, 3998–4012.
- Humme, S.; Reisbach, G.; Feederle, R.; Delecluse, H.J.; Bousset, K.; Hammerschmidt, W.; Schepers, A. The EBV nuclear antigen 1 (EBNA1) enhances B cell immortalization several thousandfold. Proc. Natl. Acad. Sci. USA 2003, 100, 10989–10994.
- Duellman, S.J.; Thompson, K.L.; Coon, J.J.; Burgess, R.R. Phosphorylation sites of Epstein-Barr virus EBNA1 regulate its function. J. Gen. Virol. 2009, 90, 2251–2259.
- Dolan, A.; Cunningham, C.; Hector, R.D.; Hassan-Walker, A.F.; Lee, L.; Addison, C.; Dargan, D.J.; McGeoch, D.J.; Gatherer, D.; Emery, V.C. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 2004, 85, 1301–1312.
- Herbein, G. The human cytomegalovirus, from oncomodulation to oncogenesis. Viruses 2018, 10, 408.
- Luo, X.H.; Meng, Q.; Rao, M.; Liu, Z.; Paraschoudi, G.; Dodoo, E.; Maeurer, M. The impact of inflationary cytomegalovirus-specific memory T cells on anti-tumour immune responses in patients with cancer. Immunology 2018, 155, 294–308.
- Lepiller, Q.; Abbas, W.; Kumar, A.; Tripathy, M.K.; Herbein, G. HCMV activates the IL-6-JAK-STAT3 axis in HepG2 cells and primary human hepatocytes. PLoS ONE 2013, 8, e59591.
- Teo, W.H.; Chen, H.-P.; Huang, J.C.; Chan, Y.-J. Human cytomegalovirus infection enhances cell proliferation, migration and upregulation of EMT markers in colorectal cancer-derived stem cell-like cells. Int. J. Oncol. 2017, 51, 1415–1426.
- Lizano, M.; Berumen, J.; García-Carrancá, A. HPV-related carcinogenesis: Basic concepts, viral types and variants. Arch. Med. Res. 2009, 40, 428–434.
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30, F55–F70.
- Jeon, S.; Allen-Hoffmann, B.L.; Lambert, P.F. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 1995, 69, 2989–2997.
- Goodwin, E.C.; Yang, E.; Lee, C.-J.; Lee, H.-W.; DiMaio, D.; Hwang, E.-S. Rapid induction of senescence in human cervical carcinoma cells. Proc. Natl. Acad. Sci. USA 2000, 97, 10978–10983.
- Shlomai, A.; de Jong, Y.P.; Rice, C.M. Virus associated malignancies: The role of viral hepatitis in hepatocellular carcinoma. Semin. Cancer Biol. 2014, 26, 78–88.
- Dandri, M.; Locarnini, S. New insight in the pathobiology of hepatitis B virus infection. Gut 2012, 61, i6–i17.
- Sukowati, C.H.; El-Khobar, K.E.; Ie, S.I.; Anfuso, B.; Muljono, D.H.; Tiribelli, C. Significance of hepatitis virus infection in the oncogenic initiation of hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 1497.
- Caldwell, S.; Park, S.H. The epidemiology of hepatocellular cancer: From the perspectives of public health problem to tumor biology. J. Gastroenterol. 2009, 44, 96–101.
- Trépo, C.; Chan, H.L.; Lok, A. Hepatitis B virus infection. Lancet 2014, 384, 2053–2063.
- Ringehan, M.; McKeating, J.A.; Protzer, U. Viral hepatitis and liver cancer. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160274.
- Castello, G.; Scala, S.; Palmieri, G.; Curley, S.A.; Izzo, F. HCV-related hepatocellular carcinoma: From chronic inflammation to cancer. Clin. Immunol. 2010, 134, 237–250.
- Wedemeyer, H. Hepatitis D revival. Liver Int. 2011, 31, 140–144.
- Knutson, K.L.; Disis, M.L.; Salazar, L.G. CD4 regulatory T cells in human cancer pathogenesis. Cancer Immunol. Immunother. 2007, 56, 271–285.
- Isaguliants, M.; Bayurova, E.; Avdoshina, D.; Kondrashova, A.; Chiodi, F.; Palefsky, J.M. Oncogenic effects of HIV-1 proteins, mechanisms behind. Cancers 2021, 13, 305.
- Katano, H. Pathological features of Kaposi’s sarcoma-associated herpesvirus infection. In Human Herpesviruses; Springer: Singapore, 2018; pp. 357–376.
- Dittmer, D.P.; Damania, B. Kaposi sarcoma–associated herpesvirus: Immunobiology, oncogenesis, and therapy. J. Clin. Investig. 2016, 126, 3165–3175.
- Rotondo, J.C.; Bononi, I.; Puozzo, A.; Govoni, M.; Foschi, V.; Lanza, G.; Gafà, R.; Gaboriaud, P.; Touzé, F.A.; Selvatici, R.; et al. Merkel Cell Carcinomas Arising in Autoimmune Disease Affected Patients Treated with Biologic Drugs, Including Anti-TNF. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 3929–3934.
- Becker, J.C.; Houben, R.; Ugurel, S.; Trefzer, U.; Pföhler, C.; Schrama, D. MC polyomavirus is frequently present in Merkel cell carcinoma of European patients. J. Investig. Dermatol. 2009, 129, 248–250.
- Verdonck, K.; González, E.; Van Dooren, S.; Vandamme, A.M.; Vanham, G.; Gotuzzo, E. Human T-lymphotropic virus 1: Recent knowledge about an ancient infection. Lancet Infect. Dis. 2007, 7, 266–281.
- Weiler, J.; Hunziker, J.; Hall, J. Anti-miRNA oligonucleotides (AMOs): Ammunition to target miRNAs implicated in human disease? Gene Ther. 2006, 13, 496–502.
- Wang, V.; Wu, W. MicroRNA-based therapeutics for cancer. BioDrugs 2009, 23, 15–23.
- Yang, X.; Marcucci, K.; Anguela, X.; Couto, L.B. Preclinical evaluation of an anti-HCV miRNA cluster for treatment of HCV infection. Mol. Ther. 2013, 21, 588–601.
- Liu, X.; Wang, T.; Wakita, T.; Yang, W. Systematic identification of microRNA and messenger RNA profiles in hepatitis C virus-infected human hepatoma cells. Virology 2010, 398, 57–67.
- Hiramatsu-Asano, S.; Wada, J. Therapeutic Approaches Targeting miRNA in Systemic Lupus Erythematosus. Acta Med. Okayama 2022, 76, 359–371.
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 2007, 4, 721–726.
- Kang, B.W.; Choi, Y.; Kwon, O.K.; Lee, S.S.; Chung, H.Y.; Yu, W.; Bae, H.I.; Seo, A.N.; Kang, H.; Lee, S.K. High level of viral microRNA-BART20-5p expression is associated with worse survival of patients with Epstein-Barr virus-associated gastric cancer. Oncotarget 2017, 8, 14988.
- Cai, L.; Li, J.; Zhang, X.; Lu, Y.; Wang, J.; Lyu, X.; Chen, Y.; Liu, J.; Cai, H.; Wang, Y. Gold nano-particles (AuNPs) carrying anti-EBV-miR-BART7-3p inhibit growth of EBV-positive nasopharyngeal carcinoma. Oncotarget 2015, 6, 7838.
- Liu, M.; Wang, W.; Chen, H.; Lu, Y.; Yuan, D.; Deng, Y.; Ran, D. miR-9, miR-21, miR-27b, and miR-34a expression in HPV16/58/52-infected cervical cancer. BioMed Res. Int. 2020, 2020, 2474235.
- Cousins, E.; Nicholas, J. Molecular Biology of Human Herpesvirus 8: Novel Functions and Virus–Host Interactions Implicated in Viral Pathogenesis and Replication. In Viruses and Human Cancer: From Basic Science to Clinical Prevention; Springer: Berlin/Heidelberg, Germany, 2014; pp. 227–268.
- Bandyopadhyay, S.; Friedman, R.C.; Marquez, R.T.; Keck, K.; Kong, B.; Icardi, M.S.; Brown, K.E.; Burge, C.B.; Schmidt, W.N.; Wang, Y. Hepatitis C virus infection and hepatic stellate cell activation downregulate miR-29: miR-29 overexpression reduces hepatitis C viral abundance in culture. J. Infect. Dis. 2011, 203, 1753–1762.
- Murakami, Y.; Aly, H.H.; Tajima, A.; Inoue, I.; Shimotohno, K. Regulation of the hepatitis C virus genome replication by miR-199a. J. Hepatol. 2009, 50, 453–460.
- Bai, X.T.; Nicot, C. miR-28-3p is a cellular restriction factor that inhibits human T cell leukemia virus, type 1 (HTLV-1) replication and virus infection. J. Biol. Chem. 2015, 290, 5381–5390.
- Wang, X.; Wang, H.-K.; McCoy, J.P.; Banerjee, N.S.; Rader, J.S.; Broker, T.R.; Meyers, C.; Chow, L.T.; Zheng, Z.-M. Oncogenic HPV infection interrupts the expression of tumor-suppressive miR-34a through viral oncoprotein E6. RNA 2009, 15, 637–647.
More
Information
Subjects:
Infectious Diseases
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
719
Revisions:
2 times
(View History)
Update Date:
19 Apr 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No