Approximately 20% of all cases of human cancer are caused by viral infections. Although a great number of viruses are capable of causing a wide range of tumors in animals, only seven of these viruses have been linked to human malignancies and are presently classified as oncogenic viruses. These include the Epstein–Barr virus (EBV), human papillomavirus (HPV), hepatitis B virus (HBV), hepatitis C virus (HCV), Merkel cell polyomavirus (MCPyV), human herpesvirus 8 (HHV8), and human T-cell lymphotropic virus type 1 (HTLV-1). It is possible that virally encoded microRNAs (miRNAs), which are ideal non-immunogenic tools for viruses, play a significant role in carcinogenic processes.
- oncogenic viruses
- viral miRNAs
- cancer
- human neoplasms
1. Oncogenic Viral Infections
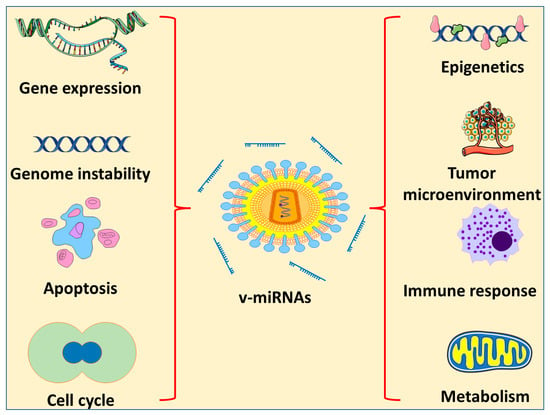
2. miRNA-Based Therapy for Oncogenic Viruses
2.1. Anti-miR
2.2. miR Mimetics
This entry is adapted from the peer-reviewed paper 10.3390/ph16040485
References
- Day, N.; Geser, A.; Lavoué, M.; Ho, J.; Simons, M.; Sohier, R.; Tukei, P.; Vonka, V.; Zavadova, H. Sero-epidemiology of the Epstein-Barr virus: Preliminary analysis of an international study—A review. IARC Sci. Publ. 1975, 11, 3–16.
- Murray, P.; Young, L. The role of the Epstein-Barr virus in human disease. Front. Biosci. Landmark 2002, 7, 519–540.
- Sixbey, J.W.; Nedrud, J.G.; Raab-Traub, N.; Hanes, R.A.; Pagano, J.S. Epstein–Barr virus replication in oropharyngeal epithelial cells. N. Engl. J. Med. 1984, 310, 1225–1230.
- Thompson, M.P.; Kurzrock, R. Epstein-Barr virus and cancer. Clin. Cancer Res. 2004, 10, 803–821.
- Mahdavifar, N.; Ghoncheh, M.; Mohammadian-Hafshejani, A.; Khosravi, B.; Salehiniya, H. Epidemiology and inequality in the incidence and mortality of nasopharynx cancer in Asia. Osong Public Health Res. Perspect. 2016, 7, 360–372.
- Chang, C.M.; Kelly, J.Y.; Mbulaiteye, S.M.; Hildesheim, A.; Bhatia, K. The extent of genetic diversity of Epstein-Barr virus and its geographic and disease patterns: A need for reappraisal. Virus Res. 2009, 143, 209–221.
- Young, L.S.; Rickinson, A.B. Epstein–Barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768.
- Yang, J.; Liu, Z.; Zeng, B.; Hu, G.; Gan, R. Epstein–Barr virus-associated gastric cancer: A distinct subtype. Cancer Lett. 2020, 495, 191–199.
- AlQarni, S.; Al-Sheikh, Y.; Campbell, D.; Drotar, M.; Hannigan, A.; Boyle, S.; Herzyk, P.; Kossenkov, A.; Armfield, K.; Jamieson, L. Lymphomas driven by Epstein–Barr virus nuclear antigen-1 (EBNA1) are dependant upon Mdm2. Oncogene 2018, 37, 3998–4012.
- Humme, S.; Reisbach, G.; Feederle, R.; Delecluse, H.J.; Bousset, K.; Hammerschmidt, W.; Schepers, A. The EBV nuclear antigen 1 (EBNA1) enhances B cell immortalization several thousandfold. Proc. Natl. Acad. Sci. USA 2003, 100, 10989–10994.
- Duellman, S.J.; Thompson, K.L.; Coon, J.J.; Burgess, R.R. Phosphorylation sites of Epstein-Barr virus EBNA1 regulate its function. J. Gen. Virol. 2009, 90, 2251–2259.
- Dolan, A.; Cunningham, C.; Hector, R.D.; Hassan-Walker, A.F.; Lee, L.; Addison, C.; Dargan, D.J.; McGeoch, D.J.; Gatherer, D.; Emery, V.C. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 2004, 85, 1301–1312.
- Herbein, G. The human cytomegalovirus, from oncomodulation to oncogenesis. Viruses 2018, 10, 408.
- Luo, X.H.; Meng, Q.; Rao, M.; Liu, Z.; Paraschoudi, G.; Dodoo, E.; Maeurer, M. The impact of inflationary cytomegalovirus-specific memory T cells on anti-tumour immune responses in patients with cancer. Immunology 2018, 155, 294–308.
- Lepiller, Q.; Abbas, W.; Kumar, A.; Tripathy, M.K.; Herbein, G. HCMV activates the IL-6-JAK-STAT3 axis in HepG2 cells and primary human hepatocytes. PLoS ONE 2013, 8, e59591.
- Teo, W.H.; Chen, H.-P.; Huang, J.C.; Chan, Y.-J. Human cytomegalovirus infection enhances cell proliferation, migration and upregulation of EMT markers in colorectal cancer-derived stem cell-like cells. Int. J. Oncol. 2017, 51, 1415–1426.
- Lizano, M.; Berumen, J.; García-Carrancá, A. HPV-related carcinogenesis: Basic concepts, viral types and variants. Arch. Med. Res. 2009, 40, 428–434.
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30, F55–F70.
- Jeon, S.; Allen-Hoffmann, B.L.; Lambert, P.F. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 1995, 69, 2989–2997.
- Goodwin, E.C.; Yang, E.; Lee, C.-J.; Lee, H.-W.; DiMaio, D.; Hwang, E.-S. Rapid induction of senescence in human cervical carcinoma cells. Proc. Natl. Acad. Sci. USA 2000, 97, 10978–10983.
- Shlomai, A.; de Jong, Y.P.; Rice, C.M. Virus associated malignancies: The role of viral hepatitis in hepatocellular carcinoma. Semin. Cancer Biol. 2014, 26, 78–88.
- Dandri, M.; Locarnini, S. New insight in the pathobiology of hepatitis B virus infection. Gut 2012, 61, i6–i17.
- Sukowati, C.H.; El-Khobar, K.E.; Ie, S.I.; Anfuso, B.; Muljono, D.H.; Tiribelli, C. Significance of hepatitis virus infection in the oncogenic initiation of hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 1497.
- Caldwell, S.; Park, S.H. The epidemiology of hepatocellular cancer: From the perspectives of public health problem to tumor biology. J. Gastroenterol. 2009, 44, 96–101.
- Trépo, C.; Chan, H.L.; Lok, A. Hepatitis B virus infection. Lancet 2014, 384, 2053–2063.
- Ringehan, M.; McKeating, J.A.; Protzer, U. Viral hepatitis and liver cancer. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160274.
- Castello, G.; Scala, S.; Palmieri, G.; Curley, S.A.; Izzo, F. HCV-related hepatocellular carcinoma: From chronic inflammation to cancer. Clin. Immunol. 2010, 134, 237–250.
- Wedemeyer, H. Hepatitis D revival. Liver Int. 2011, 31, 140–144.
- Knutson, K.L.; Disis, M.L.; Salazar, L.G. CD4 regulatory T cells in human cancer pathogenesis. Cancer Immunol. Immunother. 2007, 56, 271–285.
- Isaguliants, M.; Bayurova, E.; Avdoshina, D.; Kondrashova, A.; Chiodi, F.; Palefsky, J.M. Oncogenic effects of HIV-1 proteins, mechanisms behind. Cancers 2021, 13, 305.
- Katano, H. Pathological features of Kaposi’s sarcoma-associated herpesvirus infection. In Human Herpesviruses; Springer: Singapore, 2018; pp. 357–376.
- Dittmer, D.P.; Damania, B. Kaposi sarcoma–associated herpesvirus: Immunobiology, oncogenesis, and therapy. J. Clin. Investig. 2016, 126, 3165–3175.
- Rotondo, J.C.; Bononi, I.; Puozzo, A.; Govoni, M.; Foschi, V.; Lanza, G.; Gafà, R.; Gaboriaud, P.; Touzé, F.A.; Selvatici, R.; et al. Merkel Cell Carcinomas Arising in Autoimmune Disease Affected Patients Treated with Biologic Drugs, Including Anti-TNF. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 3929–3934.
- Becker, J.C.; Houben, R.; Ugurel, S.; Trefzer, U.; Pföhler, C.; Schrama, D. MC polyomavirus is frequently present in Merkel cell carcinoma of European patients. J. Investig. Dermatol. 2009, 129, 248–250.
- Verdonck, K.; González, E.; Van Dooren, S.; Vandamme, A.M.; Vanham, G.; Gotuzzo, E. Human T-lymphotropic virus 1: Recent knowledge about an ancient infection. Lancet Infect. Dis. 2007, 7, 266–281.
- Weiler, J.; Hunziker, J.; Hall, J. Anti-miRNA oligonucleotides (AMOs): Ammunition to target miRNAs implicated in human disease? Gene Ther. 2006, 13, 496–502.
- Wang, V.; Wu, W. MicroRNA-based therapeutics for cancer. BioDrugs 2009, 23, 15–23.
- Yang, X.; Marcucci, K.; Anguela, X.; Couto, L.B. Preclinical evaluation of an anti-HCV miRNA cluster for treatment of HCV infection. Mol. Ther. 2013, 21, 588–601.
- Liu, X.; Wang, T.; Wakita, T.; Yang, W. Systematic identification of microRNA and messenger RNA profiles in hepatitis C virus-infected human hepatoma cells. Virology 2010, 398, 57–67.
- Hiramatsu-Asano, S.; Wada, J. Therapeutic Approaches Targeting miRNA in Systemic Lupus Erythematosus. Acta Med. Okayama 2022, 76, 359–371.
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 2007, 4, 721–726.
- Kang, B.W.; Choi, Y.; Kwon, O.K.; Lee, S.S.; Chung, H.Y.; Yu, W.; Bae, H.I.; Seo, A.N.; Kang, H.; Lee, S.K. High level of viral microRNA-BART20-5p expression is associated with worse survival of patients with Epstein-Barr virus-associated gastric cancer. Oncotarget 2017, 8, 14988.
- Cai, L.; Li, J.; Zhang, X.; Lu, Y.; Wang, J.; Lyu, X.; Chen, Y.; Liu, J.; Cai, H.; Wang, Y. Gold nano-particles (AuNPs) carrying anti-EBV-miR-BART7-3p inhibit growth of EBV-positive nasopharyngeal carcinoma. Oncotarget 2015, 6, 7838.
- Liu, M.; Wang, W.; Chen, H.; Lu, Y.; Yuan, D.; Deng, Y.; Ran, D. miR-9, miR-21, miR-27b, and miR-34a expression in HPV16/58/52-infected cervical cancer. BioMed Res. Int. 2020, 2020, 2474235.
- Cousins, E.; Nicholas, J. Molecular Biology of Human Herpesvirus 8: Novel Functions and Virus–Host Interactions Implicated in Viral Pathogenesis and Replication. In Viruses and Human Cancer: From Basic Science to Clinical Prevention; Springer: Berlin/Heidelberg, Germany, 2014; pp. 227–268.
- Bandyopadhyay, S.; Friedman, R.C.; Marquez, R.T.; Keck, K.; Kong, B.; Icardi, M.S.; Brown, K.E.; Burge, C.B.; Schmidt, W.N.; Wang, Y. Hepatitis C virus infection and hepatic stellate cell activation downregulate miR-29: miR-29 overexpression reduces hepatitis C viral abundance in culture. J. Infect. Dis. 2011, 203, 1753–1762.
- Murakami, Y.; Aly, H.H.; Tajima, A.; Inoue, I.; Shimotohno, K. Regulation of the hepatitis C virus genome replication by miR-199a. J. Hepatol. 2009, 50, 453–460.
- Bai, X.T.; Nicot, C. miR-28-3p is a cellular restriction factor that inhibits human T cell leukemia virus, type 1 (HTLV-1) replication and virus infection. J. Biol. Chem. 2015, 290, 5381–5390.
- Wang, X.; Wang, H.-K.; McCoy, J.P.; Banerjee, N.S.; Rader, J.S.; Broker, T.R.; Meyers, C.; Chow, L.T.; Zheng, Z.-M. Oncogenic HPV infection interrupts the expression of tumor-suppressive miR-34a through viral oncoprotein E6. RNA 2009, 15, 637–647.