Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Cristina Maccallini | -- | 2392 | 2023-04-12 08:52:47 | | | |
| 2 | Rita Xu | Meta information modification | 2392 | 2023-04-12 10:11:23 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Amoroso, R.; Maccallini, C.; Bellezza, I. Nrf2 Activation in Neurodegenerative Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/42970 (accessed on 23 July 2026).
Amoroso R, Maccallini C, Bellezza I. Nrf2 Activation in Neurodegenerative Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/42970. Accessed July 23, 2026.
Amoroso, Rosa, Cristina Maccallini, Ilaria Bellezza. "Nrf2 Activation in Neurodegenerative Diseases" Encyclopedia, https://encyclopedia.pub/entry/42970 (accessed July 23, 2026).
Amoroso, R., Maccallini, C., & Bellezza, I. (2023, April 12). Nrf2 Activation in Neurodegenerative Diseases. In Encyclopedia. https://encyclopedia.pub/entry/42970
Amoroso, Rosa, et al. "Nrf2 Activation in Neurodegenerative Diseases." Encyclopedia. Web. 12 April, 2023.
Copy Citation
Neurodegenerative diseases are incurable and debilitating conditions that result in progressive degeneration and loss of nerve cells. Oxidative stress has been proposed as one factor that plays a potential role in the pathogenesis of neurodegenerative disorders since neuron cells are particularly vulnerable to oxidative damage. Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) is strictly related to anti-inflammatory and antioxidative cell response; therefore, its activation and the consequent enhancement of the related cellular pathways have been proposed as a potential therapeutic approach.
electrophilic activators
multitargeting compounds
Nrf2 inducers
1. Introduction
Neurodegenerative diseases affect millions of people worldwide and are a leading cause of disability and a major cause of mortality [1]. The treatment of these pathological conditions is only palliative; therefore, there is an urgent need for effective therapeutic agents, as well as a deeper understanding of the molecular changes affecting neuronal cells during the disease progression. Neuroinflammation is a hallmark in the development of neurodegenerative diseases, as well as nitroxidative stress, which is due to the unbalanced production of both reactive oxygen species (ROS) and reactive nitrogen species (RNS) [2][3]. The key role played by ROS in the onset of age-related neurodegenerative diseases indicates that erythroid-derived 2-like 2 (Nrf2), the master regulator of redox homeostasis [4], may be a promising target for therapeutic interventions. Several Nrf2 activators with different mechanisms and diverse structures have been reported in the literature, but those applied to neurodegenerative diseases are still limited [5]. However, in the very last few years, interesting progress has been made in this field, particularly in enhancing the blood–brain barrier (BBB) permeability of Nrf2 activators, to make them effective drugs, and in designing Nrf2-based multitarget-directed ligands to affect multiple pathways involved in the pathology of neurodegenerative diseases.
2. Implication of Oxidative Stress and Nrf2 Activation in Neurodegenerative Diseases
ROS are a byproduct of several cellular metabolic pathways and enzymatic reactions, and they are classified as radicals, including superoxide anion (O2●−) and hydroxyl (HO●−), as well as nonradical species such as hydrogen peroxide (H2O2) [6]. ROS are reactive molecules with a very short half-life. H2O2, the most stable ROS, has a cellular half-life of 10−3 s, 1000 times higher than other ROS [7][8]. Although ROS production might be due to environmental factors, it mainly derives from metabolic activities. During mitochondrial respiration, in the electron transport chain (ETC), approximately 1–2% of O2 is not reduced to water, leading to the generation of O2●− and H2O2. Moreover, cytosolic oxidoreductases such as NADPH oxidases (NOX), cytochrome P450 (CYP) oxidases, cyclooxygenases (COX), and monoamine oxidases (MAO) may contribute to ROS production [8]. Physiological ROS levels are maintained by a plethora of exogenous and endogenous antioxidant defenses. Among the exogenous antioxidants, ascorbic acid (vitamin C), α-tocopherol (vitamin E), and carotenoids play a pivotal role. Endogenous antioxidants include enzymatic antioxidants, e.g., superoxide dismutase (SOD), glutathione peroxidase (GPX), and catalase (CAT), as well as nonenzymatic scavengers, e.g., glutathione (GSH) (Figure 1) [9].
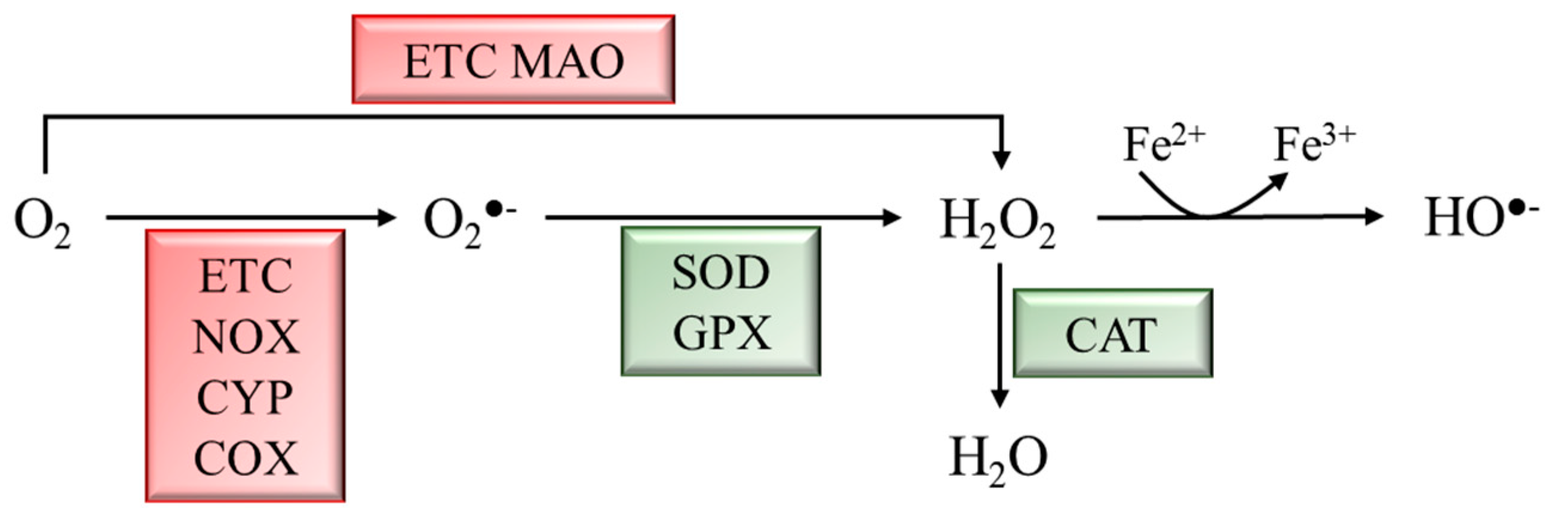
Figure 1. Schematic representation of ROS generation and scavenging. Molecular oxygen (O2) can be partially reduced to superoxide anion (O2●−) by the ETC, NADPH oxidases (NOX), cytochrome P450 (CYP) oxidases, and cyclooxygenases (COX), or to hydrogen peroxide (H2O2) by the ETC and monoamine oxidases (MAO). H2O2 can be converted into hydroxyl radical (HO●−) through the Fenton rection. Enzymatic antioxidants scavenge ROS to produce less reactive molecules. Superoxide dismutase (SOD) and glutathione peroxidase (GPX) convert O2●− to H2O2, which is converted into H2O by catalase (CAT).
Redox homeostasis should be strictly controlled since ROS play fundamental biological roles in guaranteeing redox signaling, a transduction system in which reversible electron transfer reactions involving ROS to effector target proteins culminate in the regulation of numerous physiological functions, including neuronal development and function, cellular proliferation and differentiation, and aging prevention [10]. For example, due to its ability to cross the phospholipidic bilayer of the cellular membrane, H2O2 can act in both an autocrine and a paracrine manner [7]. When the production of ROS exceeds cellular detoxification capacity, redox balance gets compromised, and oxidative stress insurges. In these conditions, high ROS levels can oxidize nucleic acids, proteins, and lipids, thus leading to cell dysfunction and eventually cell death [9][11].
The master regulator of redox homeostasis is erythroid-derived 2-like 2 (Nrf2) [4]. Nrf2 is a cap ‘n’ collar (CNC) basic leucine zipper (bZIP) transcription factor responsible for the expression of genes containing the antioxidant responsive element (ARE) sequence in their promoter region, including genes linked to the synthesis or use of GSH [12]. Under basal conditions, Nrf2 has a rapid turnover, with a half-life of approximately 20 min. In fact, Nrf2 activity is controlled by a cytoplasmic repressor protein Keap1, which sequesters Nrf2 in the cytosol and, by recruiting CUL3-dependent E3-ubiquitin ligase, leads to its ubiquitination and proteasomal degradation [13][14]. Under oxidative stress conditions, two of the 27 Cys residues in Keap1 become oxidized, causing a conformational change that, according to the “hinge and latch” model [15][16], impedes the correct orientation of Nrf2 and inhibits its ubiquitination and degradation [13]. The newly synthesized Nrf2 can in turn translocate to the nucleus to exert its functions [12]. Outside of Keap-1, Nrf2 activity is also controlled by other regulators. For example, glycogen synthase kinase-3β (GSK-3β) phosphorylates Nrf2, aiding its ubiquitination by β-transducin repeat-containing protein (β -TrCP)/Cullin-1 E3 ubiquitin ligase in a Keap-1-independent manner [17]. GSK-3β also phosphorylates the protein kinase Fyn, which translocates into the nucleus where it phosphorylates Nrf2, leading to its nuclear export and ubiquitin-dependent proteasomal degradation [18]. To add a further layer of complexity, the BTB and CNC homology transcription factors (BACH1 and BACH2) repress Nrf2 activity by competing for ARE binding [19]. On the other hand, p62/sequestosome 1 (p62/SQSTM1), a ubiquitin-binding protein, competing with Nrf2 for Keap-1 binding, leads to Nrf2 stabilization and, hence, activation (Figure 2) [20].
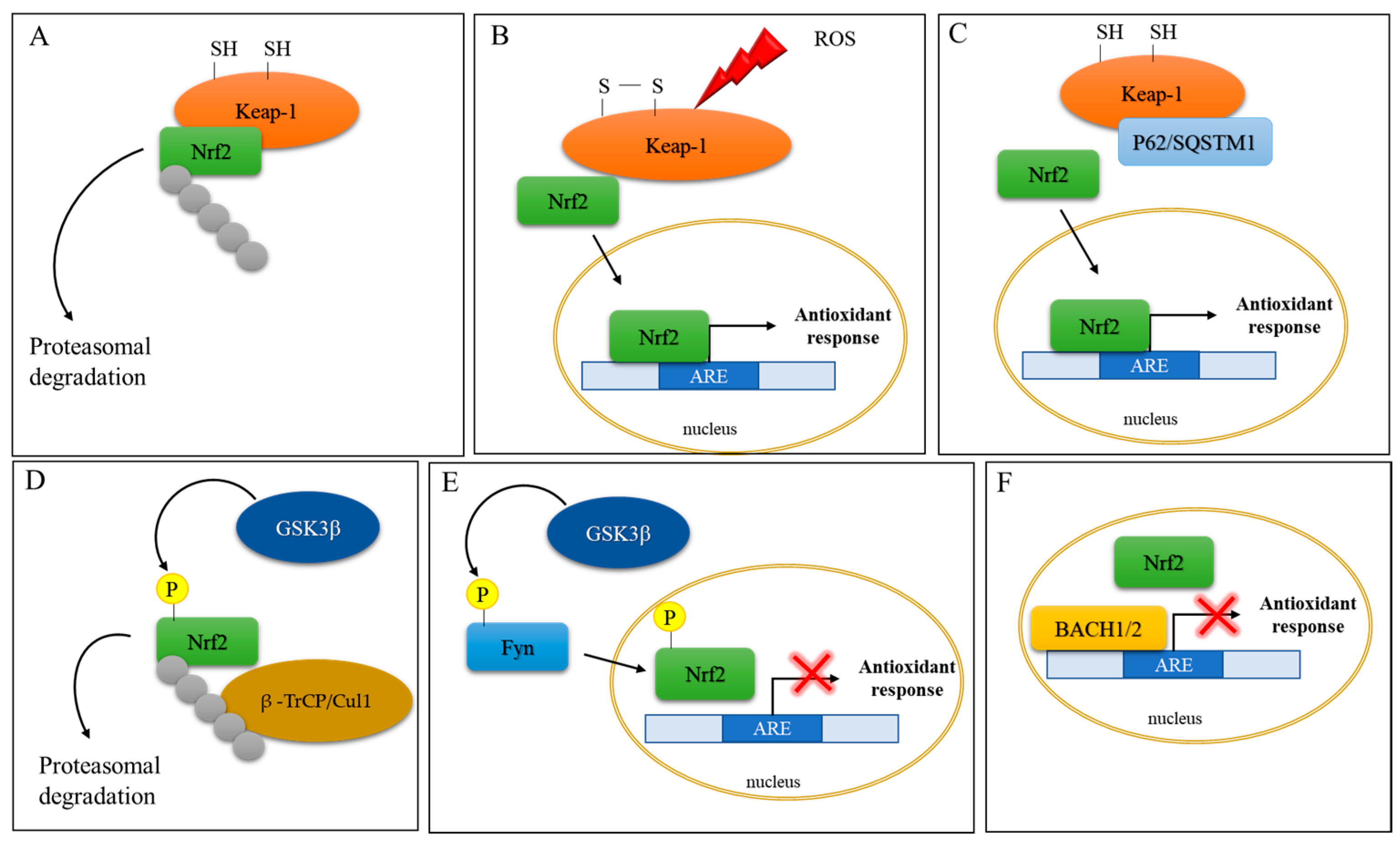
Figure 2. Schematic representation of Nrf2 activation. (A) Under basal conditions, Nrf2 activity is controlled by the cytoplasmic repressor protein Keap1, which sequesters Nrf2 in the cytosol, leading to its ubiquitination and proteasomal degradation. (B) Under oxidative stress conditions, two of the Cys residues in Keap1 become oxidized, impeding the correct orientation of Nrf2 and inhibiting its ubiquitination and degradation. The newly synthesized Nrf2 translocates to the nucleus to induce the expression of antioxidant response genes. (C) P62/sequestosome 1 (p62/SQSTM1) competes with Nrf2 for Keap-1 binding and leads to Nrf2 activation and nuclear translocation. (D) Glycogen synthase kinase-3β (GSK-3β) phosphorylates Nrf2, which is in turn ubiquitinated by β-transducin repeat-containing protein/Cullin-1 E3 ubiquitin ligase (β–TrCP/Cul1). (E) GSK-3β phosphorylates the protein kinase Fyn, which phosphorylates Nrf2, leading to its nuclear export and culminating in inhibition of gene expression (red cross). (F) The BTB and CNC homology transcription factors (BACH1 and BACH2) repress Nrf2 activity (red cross) by competing for ARE binding.
Due to the high oxygen consumption, the presence of polyunsaturated fatty acids, and auto-oxidation of neurotransmitters (e.g., dopamine), the brain is highly vulnerable to ROS-mediated damage [21]. Thus, any imbalance of redox homeostasis could affect brain cells. For example, aging and age-related diseases such as neurodegenerative disorders have been linked to the progressive dysfunction of redox control mechanisms [22]. The disruption of redox balance during healthy aging correlates with the finding that Nrf2 activity in brain decreases with age [23]. Brain expression of Nrf2 is higher in glial cells (astrocytes and microglia) than in neurons, suggesting that glial cells can be causally involved in neurodegenerative diseases despite not being the cells primarily affected by the disease. This concept, known as a non-cell-autonomous mechanism, is recognized to essentially contribute to neurodegeneration. It has been proposed that microglia and astrocytes causally participate in the pathogenesis and progression of several neurodegenerative diseases [24][25].
Microglial cells behave as the innate immune cells of the CNS [26]. Resting microglial cells have a ramified morphology; however, when activated by a danger signal, they undergo a morphological change, becoming amoeboid. While resting microglial cells continuously monitor brain activities to maintain homeostasis, activated microglial cells release cytokines and proinflammatory mediators to eliminate the threat. This acute response of microglial cells is protective, but overactivation of microglia, by inducing severe oxidative stress and neuroinflammation, leads to many neurodegenerative diseases [26]. In aged microglial cells, migration ability and phagocytosis are compromised, and the release of proinflammatory cytokines lasts for prolonged periods, strongly contributing to neurodegeneration [27]. Disturbance of Nrf2 activity and, hence, increased oxidative stress have been linked to several neurodegenerative diseases such as Parkinson’s disease and Alzheimer’s disease.
2.1. Nrf2 and Parkinson’s Disease
PD is a progressive neurological movement disorder characterized by tremor, bradykinesia, rigid muscles, and loss of postural balance [28]. The clinical feature of PD is the depletion of dopaminergic (DArgic) neurons in the substantia nigra pars compacta (SNpc), which results in dopamine (DA) deficiency [29]. PD is prevalently an idiopathic disease, with only 10–15% of cases having a family history. However, Lewy bodies found in both sporadic and familial cases of PD contain aggregated forms of α-synuclein (α-syn). The α-syn oligomers are cytotoxic to DArgic neurons [30]. The toxic effects of α-syn oligomers can be ascribed to α-syn enzymatic ferrireductase activity. Aggregation indeed inhibits α-syn enzymatic activity, leading to an accumulation of oxidized Fe, which can participate in the Fenton reaction, culminating in oxidative stress induction [31]. Moreover, mitochondrial α-syn accumulation can suppress mitochondrial respiratory complex I, leading to ROS production and, thus, to oxidative stress [32]. The crucial role of mitochondria-generated ROS in PD pathogenesis was determined from the finding that the 1-methyl-4-phenyl 1,2,3,6-tetrahydropyridine (MPTP) metabolite 1-methyl-4-phenylpyridinium (MPP+) hampers mitochondrial complex I, causing electron leakage and ROS production [33]. PD-like symptoms indeed arose in individuals using illegal drugs contaminated by MPTP, and brain postmortem examinations revealed damage to the DArgic neurons in the SNpc.
Other PD genetic risk factors comprise genes involved in the control of mitochondrial redox balance including Parkin, PTEN-induced kinase 1 (PINK1), parkinsonism-associated deglycase (PARK7), and leucine-rich repeat serine/threonine protein kinase 2 (LRRK2) [34]. Parkin, an E3 ubiquitin ligase, and PINK1, a serine/threonine kinase, form a signal transduction pathway exerting a pivotal role in the removal of damaged mitochondria by the mitophagic process [35]; PARK7 encodes for DJ-1, a protein that, by binding the mitochondrial complex I, improves its activity [36]. Mutations in PINK1, DJ-1, and LRRK2 result in mitochondrial dysfunction with impaired bioenergetics and lead to uncontrolled ROS generation [36][37]. It should be emphasized that DA metabolism itself is a source of oxidative stress. In fact, excess DA can be catabolized by monoamine oxidase (MAO), producing H2O2 or undergoing auto-oxidation to produce highly reactive dopamine quinones (DAQs) and semiquinones [38].
The crucial role played by ROS in PD indicates Nrf2 implication in this disease. In vivo studies showed that both treatment with MPTP and overexpression of α-syn increased DArgic cell death in Nrf2 KO mice [39][40]. Moreover, the Nrf2-induced cytoprotective gene NQO1 (NADPH-quinone oxidoreductase) is partially sequestered in Lewy bodies, together with p62/SQSTM1, a known activator of Nrf2 [41]. Moreover, Bach1 knockout protects mice against MPTP-induced dopaminergic cell death [42]. Moreover, the brain of LRRK2-transgenic mice and the LRRK2-overexpressing neuronal cell line show reduced expression of Nrf2 and its target genes via GSK3β activation [43]. Studies in Drosophila showed that Nrf2 activation induces mitophagy and counteracts neuronal degeneration in Parkin/Pink1 knockdown flies [44]. In addition to direct neuronal toxicity, α-syn oligomers can activate microglial cells promoting neuroinflammation, which, by further increasing ROS production via NADPH oxydase 2 (NOX2) activation, augments oxidative stress [34][41]. In this context, DJ-1, by interacting with the p47phox subunit of NOX2, inhibits its action, reducing ROS production [45]. From all the above, PD genetic risk factors are connected to aberrant Nrf2 signaling which might contribute to oxidative stress (Figure 3).
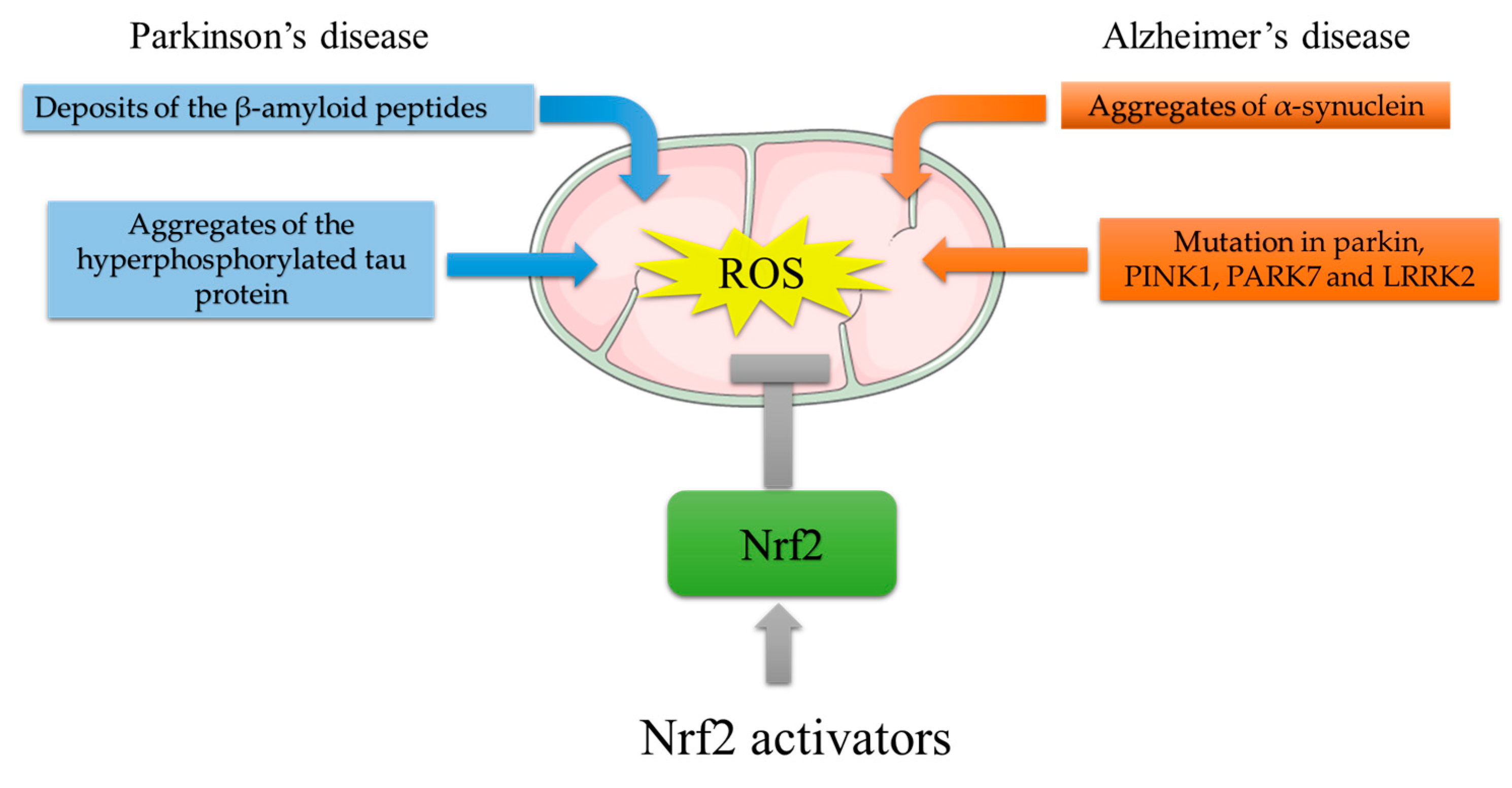
Figure 3. Mitochondrial ROS generation by PD and AD risk factors, and protective effects of Nrf2 activating compounds. ROS generation in AD: deposition of Aβ plaques induces mitochondrial stress; aggregates of the hyperphosphorylated tau protein impair mitochondria distribution and function. ROS generation in PD: aggregated α-syn accumulation suppresses mitochondrial respiratory complex I; mutations in Parkin, PTEN-induced kinase 1 (PINK1), parkinsonism-associated deglycase (PARK7), and leucine-rich repeat serine/threonine protein kinase 2 (LRRK2) result in mitochondrial dysfunction with impaired bioenergetics. The Nrf2-mediated increase in antioxidant response genes can reduce ROS burst. In this light, Nrf2 activators might counteract the ROS-induced damage that leads to neurodegeneration.
2.2. Nrf2 and Alzheimer’s Disease
Alzheimer’s disease (AD) is the prevalent type of dementia in the elderly population. AD is characterized by memory loss and deterioration of other cognitive functions including comprehension, judgment, and orientation due to synaptic loss and selective neuronal death [46]. AD brains are characterized by the accumulation of extracellular senile plaques composed of deposits of β-amyloid peptides Aβ (1–40) and Aβ (1–42), as well as intracellular neurofibrillary tangles (NFTs) composed of aggregates of the hyperphosphorylated tau protein [40]. Most AD cases are classified as sporadic late-onset AD (onset after 65 years of age), whereas only 5% are autosomal-dominant early-onset AD [47][48]. Early-onset AD is caused by a mutation in three proteins involved in the amyloidogenic pathway: APP (amyloid precursor protein), γ-secretase presenilin 1 (PSEN1), and presenilin 2 (PSEN2). The key role played by ROS in aging and the fact that idiopathic AD is an age-related disease strongly suggest that ROS are involved in AD pathogenesis. This hypothesis is corroborated by the finding that Aβ binds to mitochondrial membranes, thus affecting mitochondrial function and accelerating ROS production [49]. The pivotal role of ROS in AD is based on the findings that Aβ (1–42) induces mitochondrial stress accompanied by ROS generation in AD patients [50][51], and that Aβ plaque deposition leads to oxidative stress, which, propagating spatially over time, leads to neuronal cell death [52][53]. Not only Aβ, but also tau protein is connected to mitochondrial ROS generation in AD. For example, compared to wildtype mice, tau knockout presented reduced oxidative damage, improved memory skills, and improved mitochondrial bioenergetics [54]. Furthermore, ROS induce tau polymerization [55], and tau overexpression induces oxidative stress in vitro and in mice [56][57]. Moreover, Nrf2 activation prevents tau protein aggregation in transgenic mice expressing mutant tau protein [58]. Nrf2-KO transgenic mice carrying both mutated APP and tau died prematurely (approximately 12 months of age) and were characterized by motor deficits, neuroinflammation [59], increased oxidative stress, and high levels of Aβ and tau aggregates [60].
A decrease in expression or inactivation of the p62/SQTRSM1 gene in mice causes neurodegeneration with AD-related symptoms and induces the formation of neurofibrillary tangles [61]. GSK3β is also involved in AD pathology since its hyperactivation has been demonstrated in AD brains [62], and GSK3β-mediated phosphorylation of PSN1 reduces neuronal viability and synaptic plasticity [63]. All these data indicate that AD-critical features, i.e., Aβ and tau protein aggregation, and oxidative stress are deeply related and cooperate in the development of pathology (Figure 3).
References
- GBD 2015 Neurological Disorders Collaborator Group. Global, regional, and national burden of neurological disorders during 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017, 16, 877–897.
- Barnham, K.; Masters, C.; Bush, A. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214.
- Maccallini, C.; Amoroso, R. Targeting neuronal nitric oxide synthase as a valuable strategy for the therapy of neurological disorders. Neural Regen. Res. 2016, 11, 1731–1734.
- Boas, S.M.; Joyce, K.L.; Cowell, R.M. The Nrf2-Dependent Transcriptional Regulation of Antioxidant Defense Pathways: Relevance for Cell Type-Specific Vulnerability to Neurodegeneration and Therapeutic Intervention. Antioxidants 2021, 11, 8.
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317.
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal 2008, 10, 1343–1374.
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta 2006, 1758, 994–1003.
- Heurtaux, T.; Bouvier, D.S.; Benani, A.; Helgueta Romero, S.; Frauenknecht, K.B.M.; Mittelbronn, M.; Sinkkonen, L. Normal and Pathological Nrf2 Signalling in the Central Nervous System. Antioxidants 2022, 11, 1426.
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell. Longev. 2019, 2019, 6175804.
- Sinenko, S.A.; Starkova, T.Y.; Kuzmin, A.A.; Tomilin, A.N. Physiological Signaling Functions of Reactive Oxygen Species in Stem Cells: From Flies to Man. Front. Cell Dev. Biol. 2021, A9, 714370.
- Stadtman, E.R.; Levine, R.L. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 2003, 250, 207–218.
- Suzuki, T.; Yamamoto, M. Stress-sensing mechanisms and the physiological roles of the Keap1-Nrf2 system during cellular stress. J. Biol. Chem. 2017, 292, 16817–16824.
- Itoh, K.; Ishii, T.; Wakabayashi, N.; Yamamoto, M. Regulatory mechanisms of cellular response to oxidative stress. Free Radic. Res. 1999, 31, 319–324.
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 2004, 24, 8477–8486.
- Tong, K.I.; Kobayashi, A.; Katsuoka, F.; Yamamoto, M. Two-site substrate recognition model for the Keap1-Nrf2 system: A hinge and latch mechanism. Biol. Chem. 2006, 387, 1311–1320.
- Horie, Y.; Suzuki, T.; Inoue, J.; Iso, T.; Wells, G.; Moore, T.W.; Mizushima, T.; Dinkova-Kostova, A.T.; Kasai, T.; Kamei, T.; et al. Molecular basis for the disruption of Keap1-Nrf2 interaction via Hinge & Latch mechanism. Commun. Biol. 2021, 4, 576.
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/β-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133.
- Kanninen, K.; White, A.R.; Koistinaho, J.; Malm, T. Targeting Glycogen Synthase Kinase-3β for Therapeutic Benefit against Oxidative Stress in Alzheimer’s Disease: Involvement of the Nrf2-ARE Pathway. Int. J. Alzheimer’s Dis. 2011, 2011, 985085.
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745.
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223.
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503.
- Tan, B.L.; Norhaizan, M.E.; Liew, W.P.; Sulaiman Rahman, H. Antioxidant and Oxidative Stress: A Mutual Interplay in Age-Related Diseases. Front. Pharmacol. 2018, 9, 1162.
- Matsumaru, D.; Motohashi, H. The KEAP1-Nrf2 System in Healthy Aging and Longevity. Antioxidants 2021, 10, 1929.
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369.
- Acioglu, C.; Li, L.; Elkabes, S. Contribution of astrocytes to neuropathology of neurodegenerative diseases. Brain Res. 2021, 1758, 147291.
- Lazdon, E.; Stolero, N.; Frenkel, D. Microglia and Parkinson’s disease: Footprints to pathology. J. Neural Transm. 2020, 127, 149–158.
- Angelova, D.M.; Brown, D.R. Microglia and the aging brain: Are senescent microglia the key to neurodegeneration? J. Neurochem. 2019, 151, 676–688.
- Todorovic, M.; Wood, S.A.; Mellick, G.D. Nrf2: A modulator of Parkinson’s disease? J. Neural Transm. 2016, 123, 611–619.
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013.
- Mehra, S.; Sahay, S.; Maji, S.K. α-Synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 890–908.
- Brown, D.R. α-Synuclein as a ferrireductase. Biochem. Soc. Trans. 2013, 41, 1513–1517.
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial α-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 2010, 486, 235–239.
- Richardson, J.R.; Caudle, W.M.; Guillot, T.S.; Watson, J.L.; Nakamaru-Ogiso, E.; Seo, B.B.; Sherer, T.B.; Greenamyre, J.T.; Yagi, T.; Matsuno-Yagi, A.; et al. Obligatory role for complex I inhibition in the dopaminergic neurotoxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Toxicol. Sci. 2007, 95, 196–204.
- Chakkittukandiyil, A.; Sajini, D.V.; Karuppaiah, A.; Selvaraj, D. The principal molecular mechanisms behind the activation of Keap1/Nrf2/ARE pathway leading to neuroprotective action in Parkinson’s disease. Neurochem. Int. 2022, 156, 105325.
- Harper, J.W.; Ordureau, A.; Heo, J.M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108.
- Sai, Y.; Zou, Z.; Peng, K.; Dong, Z. The Parkinson’s disease-related genes act in mitochondrial homeostasis. Neurosci. Biobehav. Rev. 2012, 36, 2034–2043.
- Dorszewska, J.; Kowalska, M.; Prendecki, M.; Piekut, T.; Kozłowska, J.; Kozubski, W. Oxidative stress factors in Parkinson’s disease. Neural Regen. Res. 2021, 16, 1383–1391.
- Hermida-Ameijeiras, A.; Méndez-Alvarez, E.; Sánchez-Iglesias, S.; Sanmartín-Suárez, C.; Soto-Otero, R. Autoxidation and MAO-mediated metabolism of dopamine as a potential cause of oxidative stress: Role of ferrous and ferric ions. Neurochem. Int. 2004, 45, 103–116.
- Innamorato, N.G.; Jazwa, A.; Rojo, A.I.; García, C.; Fernández-Ruiz, J.; Grochot-Przeczek, A.; Stachurska, A.; Jozkowicz, A.; Dulak, J.; Cuadrado, A. Different susceptibility to the Parkinson’s toxin MPTP in mice lacking the redox master regulator Nrf2 or its target gene heme oxygenase-1. PLoS ONE 2010, 5, e11838.
- Lastres-Becker, I.; Ulusoy, A.; Innamorato, N.G.; Sahin, G.; Rábano, A.; Kirik, D.; Cuadrado, A. α-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 2012, J21, 3173–3192.
- Lastres-Becker, I.; García-Yagüe, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kügler, S.; Rábano, A.; Cuadrado, A. Repurposing the Nrf2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson’s Disease. Antioxid. Redox Signal 2016, 25, 61–77.
- Ahuja, M.; Ammal Kaidery, N.; Attucks, O.C.; McDade, E.; Hushpulian, D.M.; Gaisin, A.; Gaisina, I.; Ahn, Y.H.; Nikulin, S.; Poloznikov, A.; et al. Bach1 derepression is neuroprotective in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2111643118.
- Kawakami, F.; Imai, M.; Tamaki, S.; Ohta, E.; Kawashima, R.; Maekawa, T.; Kurosaki, Y.; Ohba, K.; Ichikawa, T. Nrf2 Expression Is Decreased in LRRK2 Transgenic Mouse Brain and LRRK2 Overexpressing SH-SY5Y Cells. Biol. Pharm. Bull. 2023, 46, 123–127.
- Gumeni, S.; Papanagnou, E.D.; Manola, M.S.; Trougakos, I.P. Nrf2 activation induces mitophagy and reverses Parkin/Pink1 knock down-mediated neuronal and muscle degeneration phenotypes. Cell Death Dis. 2021, 12, 671.
- Belarbi, K.; Cuvelier, E.; Destée, A.; Gressier, B.; Chartier-Harlin, M.C. NADPH oxidases in Parkinson’s disease: A systematic review. Mol. Neurodegener. 2017, 12, 84.
- Fu, W.Y.; Ip, N.Y. The role of genetic risk factors of Alzheimer’s disease in synaptic dysfunction. Semin. Cell Dev. Biol. 2023, 139, 3–12.
- Qiu, C.; Kivipelto, M.; von Strauss, E. Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128.
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403.
- Wang, X.; Su, B.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2009, 109 (Suppl. 1), 153–159.
- Hensley, K.; Hall, N.; Subramaniam, R.; Cole, P.; Harris, M.; Aksenov, M.; Aksenova, M.; Gabbita, S.P.; Wu, J.F.; Carney, J.M.; et al. Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J. Neurochem. 1995, 65, 2146–2156.
- Chen, Z.; Zhong, C. Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281.
- Xie, H.; Hou, S.; Jiang, J.; Sekutowicz, M.; Kelly, J.; Bacskai, B.J. Rapid cell death is preceded by amyloid plaque-mediated oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 7904–7909.
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464.
- Jara, C.; Aránguiz, A.; Cerpa, W.; Tapia-Rojas, C.; Quintanilla, R.A. Genetic ablation of tau improves mitochondrial function and cognitive abilities in the hippocampus. Redox Biol. 2018, 18, 279–294.
- Gamblin, T.-C.; King, M.-E.; Kuret, J.; Berry, R.W.; Binder, L.I. Oxidative regulation of fatty acid-induced tau polymerization. Biochemistry 2000, 39, 14203–14210.
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 2002, 156, 1051–1063.
- Cente, M.; Filipcik, P.; Pevalova, M.; Novak, M. Expression of a truncated tau protein induces oxidative stress in a rodent model of tauopathy. Eur. J. Neurosci. 2006, 24, 1085–1090.
- Stack, C.; Jainuddin, S.; Elipenahli, C.; Gerges, M.; Starkova, N.; Starkov, A.A.; Jové, M.; Portero-Otin, M.; Launay, N.; Pujol, A.; et al. Methylene blue upregulates Nrf2/ARE genes and prevents tau-related neurotoxicity. Hum. Mol. Genet. 2014, J23, 3716–3732.
- Rojo, A.I.; Pajares, M.; García-Yagüe, A.J.; Buendia, I.; Van Leuven, F.; Yamamoto, M.; López, M.G.; Cuadrado, A. Deficiency in the transcription factor Nrf2 worsens inflammatory parameters in a mouse model with combined tauopathy and amyloidopathy. Redox Biol. 2018, 18, 173–180.
- Rojo, A.I.; Pajares, M.; Rada, P.; Nuñez, A.; Nevado-Holgado, A.J.; Killik, R.; Van Leuven, F.; Ribe, E.; Lovestone, S.; Yamamoto, M.; et al. Nrf2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox Biol. 2017, 13, 444–451.
- Ramesh Babu, J.; Lamar Seibenhener, M.; Peng, J.; Strom, A.-L.; Kemppainen, R.; Cox, N.; Zhu, H.; Wooten, M.-C.; Diaz-Meco, M.-T.; Moscat, J.; et al. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J. Neurochem. 2008, 106, 107–120.
- Leroy, K.; Yilmaz, Z.; Brion, J.P. Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 2007, 33, 43–55.
- Uemura, K.; Kuzuya, A.; Shimozono, Y.; Aoyagi, N.; Ando, K.; Shimohama, S.; Kinoshita, A. GSK3beta activity modifies the localization and function of presenilin 1. J. Biol. Chem. 2007, 282, 15823–15832.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
12 Apr 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No