 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Zeljka Minic | -- | 2218 | 2023-04-11 14:54:44 | | | |
| 2 | Lindsay Dong | Meta information modification | 2218 | 2023-04-13 08:43:26 | | |
Video Upload Options
Chronic pain affects many people world-wide, and this number is continuously increasing. There is a clear link between chronic pain and the development of cardiovascular disease through activation of the sympathetic nervous system.
1. Sympathetic Nervous System and Cardiovascular Disease
2. Basic Neurocircuitry of the Sympathetic Nervous System
3. Basic Neurocircuitry of Nociception
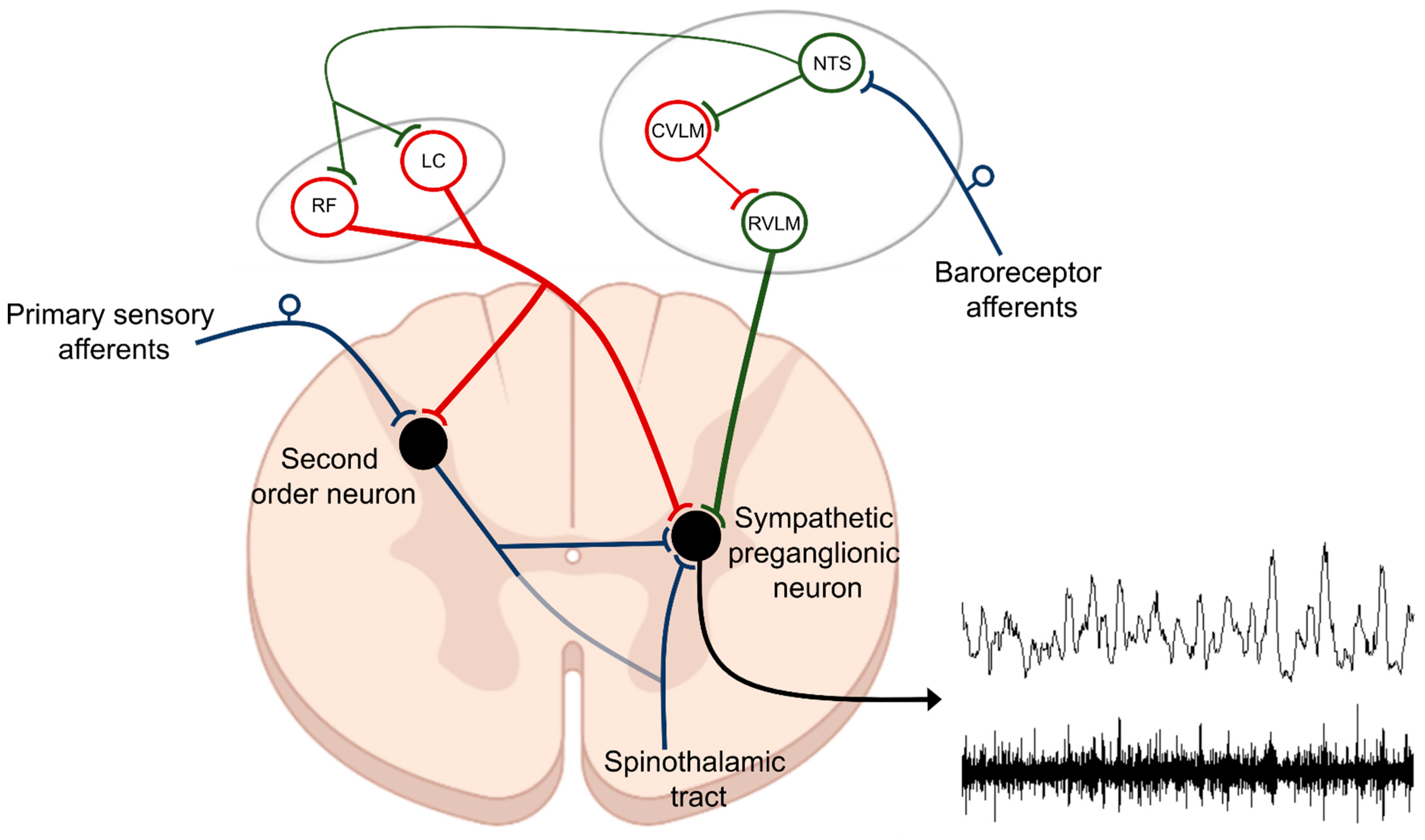
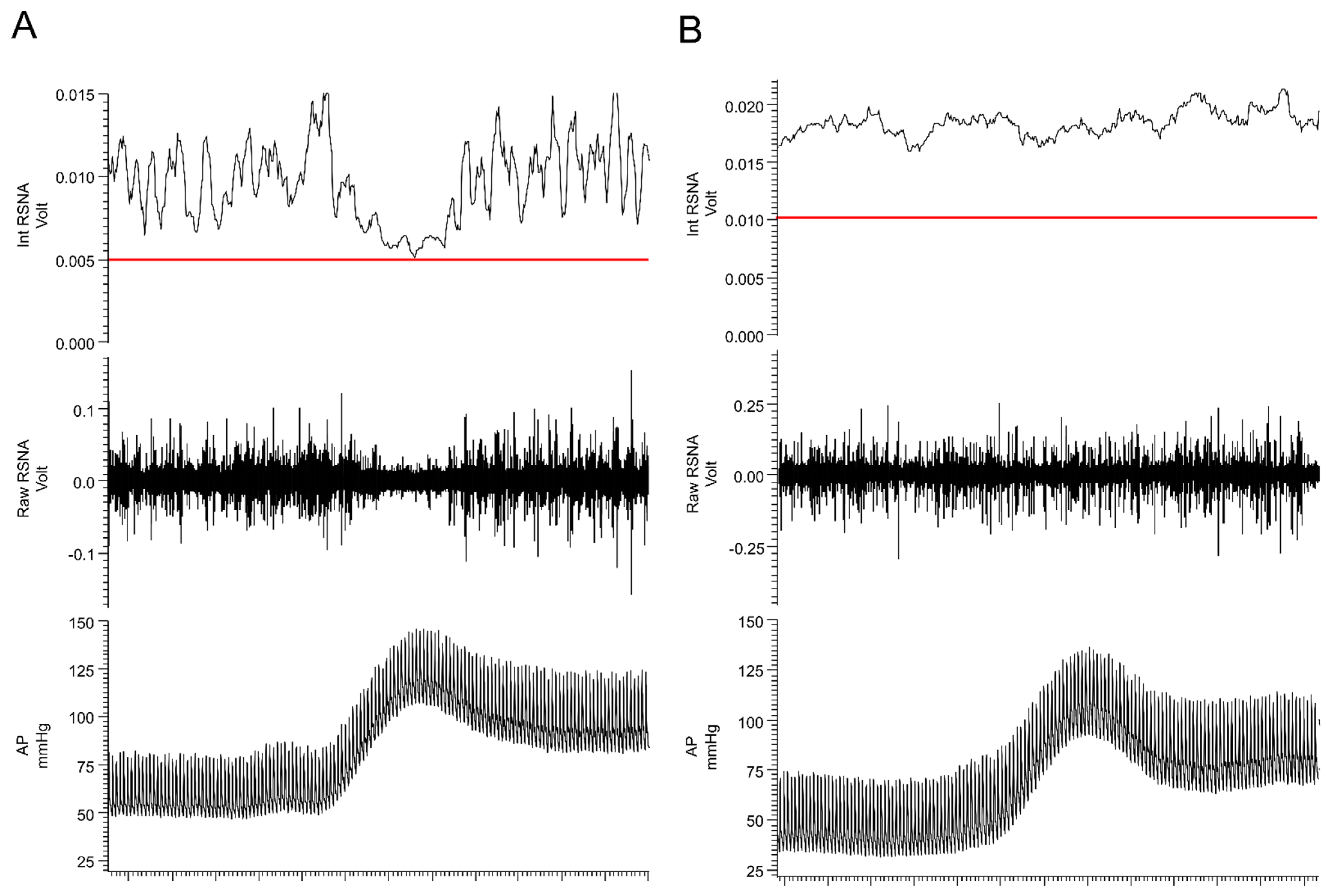
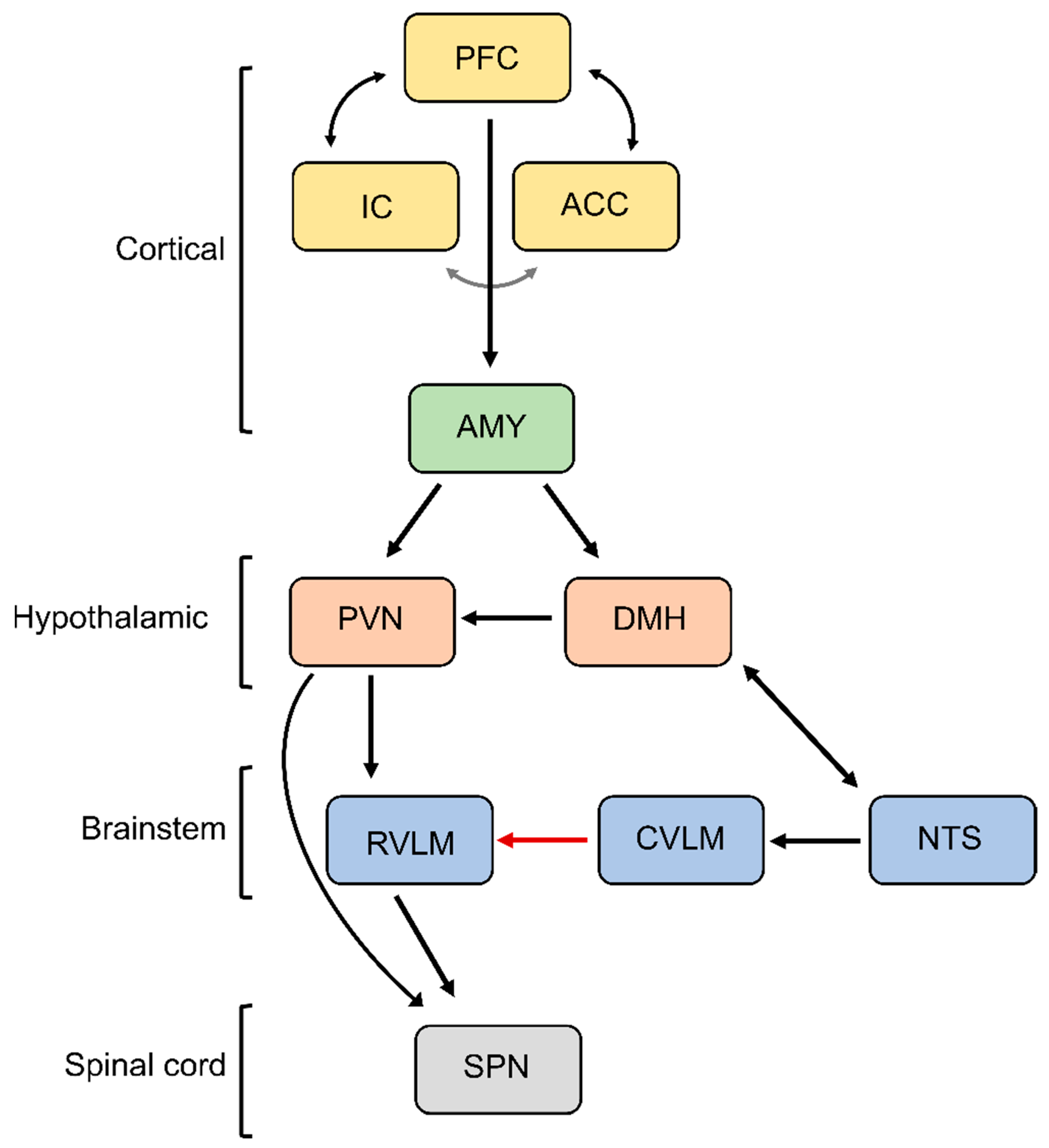
Based on the presence of thermal, chemical, or mechanical stimuli, the signal is detected by specialized receptors (nociceptors) expressed on peripheral terminals of small thinly myelinated or unmyelinated A-delta and C fibers of primary afferent neurons, respectively [40][41]. These afferents carry nociceptive information into laminae I-III of the dorsal horn of the spinal cord where they undergo complex processing. The excitatory and inhibitory interneuronal circuitry integrates incoming information and transmits it to projecting neurons for relay to the brain. Nociceptive information travels to the brain via the contralateral spinothalamic tract synapsing within the ventromedial and mediodorsal nuclei of the thalamus [42]. The nociceptive information also reaches the medulla and brainstem centers via the spinoreticular and spinomesencephalic tracts and the hypothalamus via the spinohypothalamic tract [43]. For the processing of nociceptive signals, especially important is the NTS, which integrates ascending noxious and non-noxious information and provides an interface between sensory and autonomic outflows. The NTS has dense projections with other sub-cortical brain regions such as periaqueductal gray, nucleus raphe magnus, and the hypothalamic nuclei, all of which are also involved in autonomic processing [42]. Finally, nociceptive information is subjected to processing at the cortical level where pain is conceptualized through localization and intensity discrimination. Regarding its localization, somatosensory cortices I and II are mostly responsible for determining the position of nociceptive stimulus while anterior cingulate cortex is involved in assigning an affective component to the pain [44][45].
4. The Relationship between Blood Pressure and Acute Pain Sensitivity
5. Chronic Pain as a Driver of SNS Overactivation
6. Maladaptive Changes in the Neural Circuitry Leading to Sympathetic Overactivation and Chronic Pain
References
- Malpas, S.C. Sympathetic nervous system overactivity and its role in the development of cardiovascular disease. Physiol. Rev. 2010, 90, 513–557.
- Rozanski, A.; Blumenthal, J.A.; Kaplan, J. Impact of psychological factors on the pathogenesis of cardiovascular disease and implications for therapy. Circulation 1999, 99, 2192–2217.
- Maixner, W.; Sigurdsson, A.; Fillingim, R.; Lundeen, T.; Booker, D. Regulation of acute and chronic orofacial pain. In Orofacial Pain and Temopromandibular Disorders; Raven Press: New York, NY, USA, 1995; pp. 85–102.
- Bruehl, S.; Chung, O.Y.; Ward, P.; Johnson, B.; McCubbin, J.A. The relationship between resting blood pressure and acute pain sensitivity in healthy normotensives and chronic back pain sufferers: The effects of opioid blockade. Pain 2002, 100, 191–201.
- Bruehl, S.; Burns, J.W.; McCubbin, J.A. Altered cardiovascular/pain regulatory relationships in chronic pain. Int. J. Behav. Med. 1998, 5, 63–75.
- Brody, S.; Angrilli, A.; Weiss, U.; Birbaumer, N.; Mini, A.; Veit, R.; Rau, H. Somatotosensory evoked potentials during baroreceptor stimulation in chronic low back pain patients and normal controls. Int. J. Psychophysiol. 1997, 25, 201–210.
- Perry, F.; Heller, P.H.; Kamiya, J.; Levine, J.D. Altered autonomic function in patients with arthritis or with chronic myofascial pain. Pain 1989, 39, 77–84.
- Hjemdahl, P.; Larsson, P.; Wallen, N. Effects of stress and beta-blockade on platelet function. Circulation 1991, 84, VI44–VI61.
- Ardlie, N.; Glew, G.; Schwartz, C. Influence of catecholamines on nucleotide-induced platelet aggregation. Nature 1966, 212, 415–417.
- O’brien, J. Some effects of adrenaline and anti-adrenaline compounds on platelets in vitro and in vivo. Nature 1963, 200, 763–764.
- Benarroch, E.E. The central autonomic network: Functional organization, dysfunction, and perspective. In Proceedings of Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 1993; pp. 988–1001.
- Calaresu, F.; Yardley, C. Medullary basal sympathetic tone. Annu. Rev. Physiol. 1988, 50, 511–524.
- Guertzenstein, P.; Silver, A. Fall in blood pressure produced from discrete regions of the ventral surface of the medulla by glycine and lesions. J. Physiol. 1974, 242, 489–503.
- Stein, R.; Weaver, L.; Yardley, C. Ventrolateral medullary neurones: Effects on magnitude and rhythm of discharge of mesenteric and renal nerves in cats. J. Physiol. 1989, 408, 571–586.
- Guyenet, P.G.; Haselton, J.R.; Sun, M.-K. Sympathoexcitatory neurons of the rostroventrolateral medulla and the origin of the sympathetic vasomotor tone. Prog. Brain Res. 1989, 81, 105–116.
- Sakima, A.; Yamazato, M.; Sesoko, S.; Muratani, H.; Fukiyama, K. Cardiovascular and sympathetic effects of L-glutamate and glycine injected into the rostral ventrolateral medulla of conscious rats. Hypertens. Res. 2000, 23, 633–641.
- Willette, R.; Barcas, P.; Krieger, A.; Sapru, H.N. Vasopressor and depressor areas in the rat medulla: Identification by microinjection of L-glutamate. Neuropharmacology 1983, 22, 1071–1079.
- Dampney, R.; Moon, E.A. Role of ventrolateral medulla in vasomotor response to cerebral ischemia. Am. J. Physiol.-Heart Circ. Physiol. 1980, 239, H349–H358.
- Sun, M.-K.; Reis, D.J. Central neural mechanisms mediating excitation of sympathetic neurons by hypoxia. Prog. Neurobiol. 1994, 44, 197–219.
- Kubo, T.; Goshima, Y.; Hata, H.; Misu, Y. Evidence that endogenous catecholamines are involved inα2-adrenoceptor-mediated modulation of the aortic baroreceptor reflex in the nucleus tractus solitarii of the rat. Brain Res. 1990, 526, 313–317.
- Lawrence, A.J.; Jarrott, B. Neurochemical modulation of cardiovascular control in the nucleus tractus solitarius. Prog. Neurobiol. 1996, 48, 21–53.
- Murase, S.; Inui, K.; Nosaka, S. Baroreceptor inhibition of the locus coeruleus noradrenergic neurons. Neuroscience 1994, 61, 635–643.
- Morrison, S.F. RVLM and raphe differentially regulate sympathetic outflows to splanchnic and brown adipose tissue. Am. J. Physiol. 1999, 276, R962–R973.
- Reynolds, C.A.; O’Leary, D.S.; Ly, C.; Smith, S.A.; Minic, Z. Development of a decerebrate model for investigating mechanisms mediating viscero-sympathetic reflexes in the spinalized rat. Am. J. Physiol.-Heart Circ. Physiol. 2019, 316, H1332–H1340.
- Mayorov, D.N.; Adams, M.A.; Krassioukov, A.V. Telemetric blood pressure monitoring in conscious rats before and after compression injury of spinal cord. J. Neurotrauma 2001, 18, 727.
- Osborn, J.W.; Taylor, R.F.; Schramm, L.P. Determinants of arterial pressure after chronic spinal transection in rats. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 1989, 256, R666–R673.
- Hayes, K.; Yardley, C.; Weaver, L. Evidence for descending tonic inhibition specifically affecting sympathetic pathways to the kidney in rats. J. Physiol. 1991, 434, 295–306.
- Fontes, M.; Tagawa, T.; Polson, J.; Cavanagh, S.-J.; Dampney, R. Descending pathways mediating cardiovascular response from dorsomedial hypothalamic nucleus. Am. J. Physiol.-Heart Circ. Physiol. 2001, 280, H2891–H2901.
- Cao, W.H.; Fan, W.; Morrison, S.F. Medullary pathways mediating specific sympathetic responses to activation of dorsomedial hypothalamus. Neuroscience 2004, 126, 229–240.
- Martin, D.S.; Segura, T.; Haywood, J.R. Cardiovascular responses to bicuculline in the paraventricular nucleus of the rat. Hypertension 1991, 18, 48–55.
- Chau, D.; Johns, D.G.; Schramm, L.P. Ongoing and stimulus-evoked activity of sympathetically correlated neurons in the intermediate zone and dorsal horn of acutely spinalized rats. J. Neurophysiol. 2000, 83, 2699–2707.
- Barman, S.M.; Gebber, G.L. Spinal interneurons with sympathetic nerve-related activity. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 1984, 247, R761–R767.
- Sherrington, C.; Laslett, E. Observations on some spinal reflexes and the interconnection of spinal segments. J. Physiol. 1903, 29, 58.
- Miller, F.R. Viscero-motor reflexes. II. Am. J. Physiol.-Leg. Content 1924, 71, 84–89.
- Miller, F.R.; Waud, R. Viscero-motor reflexes. IV. Am. J. Physiol.-Leg. Content 1925, 73, 329–340.
- Downman, C.; McSwiney, B. Reflexes elicited by visceral stimulation in the acute spinal animal. J. Physiol. 1946, 105, 80.
- Minic, Z.; O’Leary, D.S.; Reynolds, C.A. Spinal reflex control of arterial blood pressure: The role of TRP channels and their endogenous eicosanoid modulators. Front. Physiol. 2022, 13, 838175.
- Jankowska, E.; Padel, Y.; Tanaka, R. Disynaptic inhibition of spinal motoneurones from the motor cortex in the monkey. J. Physiol. 1976, 258, 467–487.
- Triggs, W.J.; Macdonell, R.A.; Cros, D.; Chiappa, K.H.; Shahani, B.T.; Day, B.J. Motor inhibition and excitation are independent effects of magnetic cortical stimulation. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1992, 32, 345–351.
- Bennett, G.J. Update on the neurophysiology of pain transmission and modulation: Focus on the NMDA-receptor. J. Pain Symptom Manag. 2000, 19, 2–6.
- Sandkühler, J.; Bromm, B.; Gebhart, G.F. Nervous System Plasticity and Chronic Pain; Elsevier Science Limited: Amsterdam, The Netherlands, 2000; Volume 129.
- Craig, A.D. Pain mechanisms: Labeled lines versus convergence in central processing. Annu. Rev. Neurosci. 2003, 26, 1–30.
- Brooks, J.; Tracey, I. From nociception to pain perception: Imaging the spinal and supraspinal pathways. J. Anat. 2005, 207, 19–33.
- Bushnell, M.C.; Duncan, G.H.; Hofbauer, R.K.; Ha, B.; Chen, J.I.; Carrier, B. Pain perception: Is there a role for primary somatosensory cortex? Proc. Natl. Acad. Sci. USA 1999, 96, 7705–7709.
- Rainville, P.; Duncan, G.H.; Price, D.D.; Carrier, B.; Bushnell, M.C. Pain affect encoded in human anterior cingulate but not somatosensory cortex. Science 1997, 277, 968–971.
- Bruehl, S.; Carlson, C.R.; McCubbin, J.A. The relationship between pain sensitivity and blood pressure in normotensives. Pain 1992, 48, 463–467.
- Bruehl, S.; Chung, O.Y. Interactions between the cardiovascular and pain regulatory systems: An updated review of mechanisms and possible alterations in chronic pain. Neurosci. Biobehav. Rev. 2004, 28, 395–414.
- McCubbin, J.A.; Bruehl, S. Do endogenous opioids mediate the relationship between blood pressure and pain sensitivity in normotensives? Pain 1994, 57, 63–67.
- Fillingim, R.B.; Maixner, W. The influence of resting blood pressure and gender on pain responses. Psychosom. Med. 1996, 58, 326–332.
- Pfleeger, M.; Straneva, P.A.; Fillingim, R.B.; Maixner, W.; Girdler, S.S. Menstrual cycle, blood pressure and ischemic pain sensitivity in women: A preliminary investigation. Int. J. Psychophysiol. 1997, 27, 161–166.
- Fillingim, R.B.; Maixner, W.; Bunting, S.; Silva, S. Resting blood pressure and thermal pain responses among females: Effects on pain unpleasantness but not pain intensity. Int. J. Psychophysiol. 1998, 30, 313–318.
- Myers, C.D.; Robinson, M.E.; Riley III, J.L.; Sheffield, D. Sex, gender, and blood pressure: Contributions to experimental pain report. Psychosom. Med. 2001, 63, 545–550.
- Al’Absi, M.; Buchanan, T.; Lovallo, W.R. Pain perception and cardiovascular responses in men with positive parental history for hypertension. Psychophysiology 1996, 33, 655–661.
- Al’Absi, M.; Buchanan, T.W.; Marrero, A.; Lovallo, W.R. Sex differences in pain perception and cardiovascular responses in persons with parental history for hypertension. Pain 1999, 83, 331–338.
- al’Absi, M.; Petersen, K.L.; Wittmers, L.E. Blood pressure but not parental history for hypertension predicts pain perception in women. Pain 2000, 88, 61–68.
- al’Absi, M.; Petersen, K.L.; Wittmers, L.E. Adrenocortical and hemodynamic predictors of pain perception in men and women. Pain 2002, 96, 197–204.
- Randich, A.; Maixner, W. Interactions between cardiovascular and pain regulatory systems. Neurosci. Biobehav. Rev. 1984, 8, 343–367.
- Takeda, M.; Tanimoto, T.; Ojima, K.; Matsumoto, S. Suppressive effect of vagal afferents on the activity of the trigeminal spinal neurons related to the jaw-opening reflex in rats: Involvement of the endogenous opioid system. Brain Res. Bull. 1998, 47, 49–56.
- Bossut, D.; Maixner, W. Effects of cardiac vagal afferent electrostimulation on the responses of trigerninal and trigeminothalamic neurons to noxious orofacial stimulation. Pain 1996, 65, 101–109.
- Dworkin, B.; Filewich, R.; Miller, N.; Craigmyle, N.; Pickering, T. Baroreceptor activation reduces reactivity to noxious stimulation: Implications for hypertension. Science 1979, 205, 1299–1301.
- Thurston, C.L.; Randich, A. Acute increases in arterial blood pressure produced by occlusion of the abdominal aorta induces antinociception: Peripheral and central substrates. Brain Res. 1990, 519, 12–22.
- Maixner, W.; Touw, K.B.; Brody, M.J.; Gebhart, G.F. Factors influencing the altered pain perception in the spontaneously hypertensive rat. Brain Res. 1982, 237, 137–145.
- D’Antono, B.; Ditto, B.; Sita, A.; Miller, S.B. Cardiopulmonary baroreflex stimulation and blood pressure-related hypoalgesia. Biol. Psychol. 2000, 53, 217–231.
- Dworkin, B.R.; Elbert, T.; Rau, H.; Birbaumer, N.; Pauli, P.; Droste, C.; Brunia, C. Central effects of baroreceptor activation in humans: Attenuation of skeletal reflexes and pain perception. Proc. Natl. Acad. Sci. USA 1994, 91, 6329–6333.
- Rau, H.; Brody, S.; Larbig, W.; Pauli, P.; Vöhringer, M.; Harsch, B.; Kröling, P.; Birbaumer, N. Effects of PRES baroreceptor stimulation on thermal and mechanical pain threshold in borderline hypertensives and normotensives. Psychophysiology 1994, 31, 480–485.
- Angrilli, A.; Mini, A.; Mucha, R.F.; Rau, H. The influence of low blood pressure and baroreceptor activity on pain responses. Physiol. Behav. 1997, 62, 391–397.
- Edwards, L.; McIntyre, D.; Carroll, D.; Ring, C.; France, C.R.; Martin, U. Effects of artificial and natural baroreceptor stimulation on nociceptive responding and pain. Psychophysiology 2003, 40, 762–769.
- Edwards, L.; Ring, C.; McIntyre, D.; Carroll, D. Modulation of the human nociceptive flexion reflex across the cardiac cycle. Psychophysiology 2001, 38, 712–718.
- Browne, T.J.; Hughes, D.I.; Dayas, C.V.; Callister, R.J.; Graham, B.A. Projection neuron axon collaterals in the dorsal horn: Placing a new player in spinal cord pain processing. Front. Physiol. 2020, 11, 560802.
- Schramm, L.P. Spinal sympathetic interneurons: Their identification and roles after spinal cord injury. Prog. Brain Res. 2006, 152, 27–37.
- Clifton, G.L.; Ziegler, M.G.; Grossman, R.G. Circulating catecholamines and sympathetic activity after head injury. Neurosurgery 1981, 8, 10–14.
- Park, E.S.; Park, C.I.; Cho, S.R.; Lee, J.W.; Kim, E.J. Assessment of autonomic nervous system with analysis of heart rate variability in children with spastic cerebral palsy. Yonsei Med. J. 2002, 43, 65–72.
- Kushki, A.; Drumm, E.; Mobarak, M.P.; Tanel, N.; Dupuis, A.; Chau, T.; Anagnostou, E. Investigating the autonomic nervous system response to anxiety in children with autism spectrum disorders. PLoS ONE 2013, 8, e59730.
- Appelhans, B.M.; Luecken, L.J. Heart rate variability as an index of regulated emotional responding. Rev. Gen. Psychol. 2006, 10, 229.
- Webb, S.; Adgey, A.; Pantridge, J. Autonomic disturbance at onset of acute myocardial infarction. Br. Med. J. 1972, 3, 89–92.
- Schwartz, P.J.; Vanoli, E. Cardiac arrhythmias elicited by interaction between acute myocardial ischemia and sympathetic hyperactivity: A new experimental model for the study of antiarrhythmic drugs. J. Cardiovasc. Pharmacol. 1981, 3, 1251–1259.
- Lyon, A.R.; Rees, P.S.; Prasad, S.; Poole-Wilson, P.A.; Harding, S.E. Stress (Takotsubo) cardiomyopathy—A novel pathophysiological hypothesis to explain catecholamine-induced acute myocardial stunning. Nat. Rev. Cardiol. 2008, 5, 22.
- Esler, M.; Lambert, G.; Brunner-La Rocca, H.; Vaddadi, G.; Kaye, D. Sympathetic nerve activity and neurotransmitter release in humans: Translation from pathophysiology into clinical practice. Acta Physiol. Scand. 2003, 177, 275–284.
- Schadt, J.C.; Ludbrook, J. Hemodynamic and neurohumoral responses to acute hypovolemia in conscious mammals. Am. J. Physiol. -Heart Circ. Physiol. 1991, 260, H305–H318.
- Evans, R.G.; Ventura, S.; Dampney, R.A.; Ludbrook, J. John Ludbrook APPS Symposium Neural Mechanisms In The Cardiovascular Responses To Acute Central Hypovolaemia. Clin. Exp. Pharmacol. Physiol. 2001, 28, 479–487.
- Carlson, C.R.; Okeson, J.P.; Falace, D.A.; Nitz, A.J.; Curran, S.L.; Anderson, D. Comparison of psychologic and physiologic functioning between patients with masticatory muscle pain and matched controls. J. Orofac. Pain 1993, 7.
- Schweinhardt, P.; Bushnell, M.C. Pain imaging in health and disease—How far have we come? J. Clin. Investig. 2010, 120, 3788–3797.
- May, A. Chronic pain may change the structure of the brain. PAIN® 2008, 137, 7–15.
- Hubbard, C.S.; Khan, S.A.; Keaser, M.L.; Mathur, V.A.; Goyal, M.; Seminowicz, D.A. Altered brain structure and function correlate with disease severity and pain catastrophizing in migraine patients. Eneuro 2014, 1.

