Chronic pain affects many people world-wide, and this number is continuously increasing. There is a clear link between chronic pain and the development of cardiovascular disease through activation of the sympathetic nervous system.
1. Sympathetic Nervous System and Cardiovascular Disease
The SNS is critical for general cardiovascular homeostasis [
39] as it exerts direct actions on heart rate and cardiac contractility as well as venous capacitance, arteriolar resistance, and blood volume (via sodium and water balance in the kidneys). However, excessive SNS activation increases cardiovascular morbidity and mortality [
39,
40]. Acute overactivation of the SNS can manifest as adverse cardiac events including: ventricular arrhythmias, myocardial infarction, atrial fibrillation, stroke, and Takotsubo cardiomyopathy [
40]. Chronic overactivation of the SNS leads to hypertension, ischemic heart disease, heart failure, and renal failure [
39].
In chronic pain, cardiovascular indices that indicate SNS overactivation, including increased blood pressure and heart rate, have been well documented [
41,
42,
43,
44,
45]. Additionally, SNS overactivation contributes to the process of atherosclerosis by inducing platelet activation [
46,
47,
48] and promoting mechanical injury to the vascular endothelial cells because of increased blood pressure and flow velocity. At the level of the heart, the ensuing atherosclerosis manifests with coronary artery disease and can trigger myocardial infarction. Furthermore, chronic, and excessive sympathetic drive to the heart (i) limits myocardial oxygen delivery via coronary vasoconstriction, while also (ii) increasing myocardial oxygen demand because of increased energy utilization.
2. Basic Neurocircuitry of the Sympathetic Nervous System
Sympathetic preganglionic neurons are cholinergic neurons located within the intermediolateral (IML) cell column of the spinal cord. These neurons innervate pre- and para-vertebral ganglia where they synapse with adrenergic postganglionic neurons. One exception is the adrenal gland, which is directly innervated by sympathetic preganglionic neurons and drives the systemic release of epinephrine. The regulation of sympathetic preganglionic neuron-firing is influenced by various interconnected neuronal networks, including: (i) the central autonomic network providing descending projections from the brain, and (ii) the intraspinal network consisting of propriospinal neurons and spinal interneurons. The level of sympathetic activity targeting the effector organs is ultimately determined by the balance of excitatory and inhibitory inputs to sympathetic preganglionic neurons in the IML.
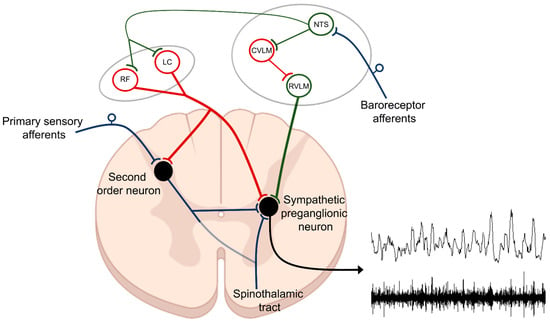
The central autonomic network serves an essential physiological role in the coordinated, real-time adaptations of SNS in response to external and internal stimuli. Brain regions, which comprise the central autonomic network include: (i) brainstem centers (both medulla and midbrain), (ii) diencephalon, and (iii) cortical sites; all of which work in a coordinated manner to influence sympathetic tone and cardiovascular function [
50]. Arguably, to date, most is known about the role of brainstem centers in controlling SNA. It was over 100 years ago when the rostral ventrolateral medulla (RVLM) was first identified as a primary regulator of sympathetic tone [
51,
52] as bilateral lesioning or pharmacological blockade of the ventrolateral medulla produced decreases in sympathetic activity and arterial hypotension [
51,
52,
53]. Since then, many studies have substantiated this initial observation and highlighted the RVLM as one of the most important sources of descending excitatory drive to sympathetic preganglionic neurons located in the IML [
54,
55,
56,
57,
58]. Additionally, the RVLM and adjacent brainstem centers are involved in baroreflex processing, which is critical for quick reflex-mediated changes in SNA (
Figure 1). Afferent neurons innervating the carotid sinus and aortic arch carry afferent, baroreceptor-related information, to the medulla, via the vagal and glossopharyngeal nerves. During baroreceptor loading, as occurs during increases in blood pressure, the nucleus of the solitary tract (NTS) receives excitatory glutamatergic input from these afferent neurons and activates inhibitory GABAergic neurons within the caudal ventrolateral medulla (CVLM). The CVLM, in turn, inhibits RVLM neurons and decreases the excitatory drive to the sympathetic preganglionic neurons located in the IML. It is important to note that in addition to decreasing excitatory RVLM drive, baroreceptor loading increases descending inhibitory drive to sympathetic preganglionic neurons located in the IML [
59,
60,
61]. Such descending inhibitory projections may arise from nuclei of the CVLM, the reticular formation, or the locus coeruleus (LC), which also receive direct projections from barosensitive NTS neurons. While the central neural networks involved in driving descending inhibition of IML neurons are not well defined, in response to baroreceptor loading, SNA can be reduced to levels similar to those observed following ganglionic blockade (
Figure 2, panel A), which is well below the levels recorded following bilateral pharmacological inhibition of RVLM [
62] or spinal cord transection (
Figure 2, panel B) [
63,
64,
65,
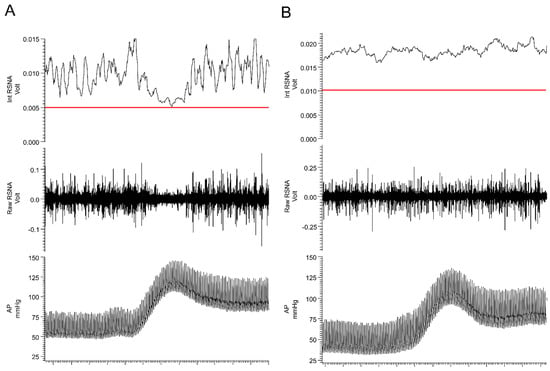
66]. These data suggest that: (i) intraspinal networks alone can maintain baseline SNA tone and blood pressure and that (ii) the inhibition of SNA in response to baroreceptor loading (phenylephrine infusion) originates from within the supraspinal centers, possibly the LC or other brainstem centers.
Figure 1. Control of sympathetic preganglionic neurons. Efferent sympathetic nerve activity (SNA) is determined by the net product of excitatory and inhibitory projections to sympathetic preganglionic neurons located within the intermediolateral (IML) cell column of the thoracic spinal cord. The rostral ventrolateral medulla (RVLM) is one of the main sources of descending excitatory drive (green). Additionally, second-order sensory neurons, relaying information from primary sensory afferents, are an important intraspinal source of excitatory drive to sympathetic preganglionic neurons. Descending inhibitory projections to sympathetic preganglionic neurons (red) arise from multiple brain regions including from the locus coeruleus (LC) and the reticular formation (RF). Much of the descending excitatory and inhibitory drive to sympathetic preganglionic neurons is regulated by neurons located within the nucleus of the solitary tract (NTS), which is the primary integrative site for baroreceptor and chemoreceptor afferent fibers which drive autonomic reflexes. Included above is an example of raw (lower tracing) and integrated (upper tracing) SNA recorded from postganglionic renal sympathetic nerve fibers in a rat as described in [
63] (lower right).
Figure 2. Baroreceptor loading strongly inhibits sympathetic nerve activity. Responses in arterial pressure (AP) and renal sympathetic nerve activity (RSNA) to intravenous administration of phenylephrine (40 µg/kg) in (A) spinal intact and (B) T4 spinal-transected rats. The red line in the top panel represents the level of recorded activity (zero) at the end of the experiment following ganglionic blockade (hexamethonium, 20 mg/kg). See text for additional information.
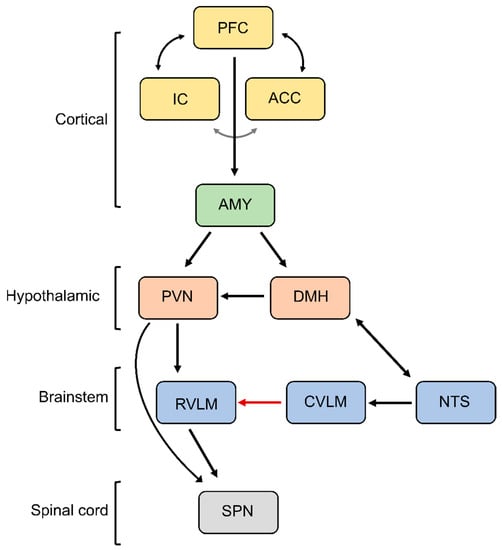
In addition to reflex regulation, cognitive processing and emotion regulation are important drivers of sympathetic activity involving the central autonomic network. Classical cardiovascular responses to emotional stress (fight and flight response or defense reaction) are produced by disinhibition of the paraventricular and dorsomedial nuclei of the hypothalamus and drive sympathoexcitatory responses and increases in blood pressure (
Figure 3) [
67,
68,
69].
Figure 3. Central autonomic network. Depicted is the neural circuitry controlling SNA during emotional stress. PFC, prefrontal cortex; IC, insular cortex; ACC, anterior cingulate cortex; AMY, amygdala; DMH, dorsomedial hypothalamus; PVN, paraventricular nucleus; RVLM, rostral ventrolateral medulla; CVLM, caudal ventrolateral medulla; NTS, nucleus of the solitary tract; SPN, sympathetic preganglionic neurons. See text for additional information.
In addition to the descending excitatory and inhibitory projections controlling sympathetic activity outlined above, sympathetic preganglionic neurons within the IML receive extensive input from intraspinal networks [
85,
86]. Spinal circuits can contribute both to baseline and reflex responses in sympathetic activity. Resting sympathetic activity and blood pressure are maintained (albeit at a lower level), following cervical or thoracic spinal cord transection and they can be further reduced by ganglionic blockade [
63,
64]. Although tonically low, sympathetic activity can be markedly increased following the activation of spinal sympathetic pathways. The spinal sympathetic reflex circuitry is similar in composition to the spinal motor reflex circuits that drive muscle contraction in response to muscle stretch or cutaneous stimulation, and like spinal motor reflexes, the spinal sympathetic reflexes are exaggerated by spinal cord transection [
87,
88,
89,
90,
91]. The spinal motoneurons are controlled by the descending corticothalamic projections, which exhibit a tonic inhibitory drive to the motoneurons [
92,
93].
3. Basic Neurocircuitry of Nociception
Based on the presence of thermal, chemical, or mechanical stimuli, the signal is detected by specialized receptors (nociceptors) expressed on peripheral terminals of small thinly myelinated or unmyelinated A-delta and C fibers of primary afferent neurons, respectively [106,107]. These afferents carry nociceptive information into laminae I-III of the dorsal horn of the spinal cord where they undergo complex processing. The excitatory and inhibitory interneuronal circuitry integrates incoming information and transmits it to projecting neurons for relay to the brain. Nociceptive information travels to the brain via the contralateral spinothalamic tract synapsing within the ventromedial and mediodorsal nuclei of the thalamus [108]. The nociceptive information also reaches the medulla and brainstem centers via the spinoreticular and spinomesencephalic tracts and the hypothalamus via the spinohypothalamic tract [109]. For the processing of nociceptive signals, especially important is the NTS, which integrates ascending noxious and non-noxious information and provides an interface between sensory and autonomic outflows. The NTS has dense projections with other sub-cortical brain regions such as periaqueductal gray, nucleus raphe magnus, and the hypothalamic nuclei, all of which are also involved in autonomic processing [108]. Finally, nociceptive information is subjected to processing at the cortical level where pain is conceptualized through localization and intensity discrimination. Regarding its localization, somatosensory cortices I and II are mostly responsible for determining the position of nociceptive stimulus while anterior cingulate cortex is involved in assigning an affective component to the pain [110,111].
4. The Relationship between Blood Pressure and Acute Pain Sensitivity
In pain-free individuals, there is a linear, inverse (negative) relationship between blood pressure and acute pain sensitivity [
143,
144,
145,
146,
147,
148,
149,
150,
151,
152,
153]. Importantly, the diminished acute pain sensitivity association with elevated resting blood pressure involves the medullary baroreflex circuitry, reducing both SNA and pain processing in response to increases in systemic blood pressure. In animals, baroreceptor stimulation induces antinociception [
154,
155,
156], and surgical denervation of baroreceptor afferents produces hyperalgesia [
157,
158,
159]. In humans, stimulation of baroreceptors (e.g., via application of external suction to the carotid artery) reduces acute pain sensitivity [
160,
161,
162,
163]. Consistent with the notion that baroreceptor loading is greatest during systole and lowest during diastole, pain sensitivity to ultra-rapid electrical pain stimuli was found to be lowest during systole and the greatest during diastole [
164,
165]. Altogether, these studies indicate that in addition to reducing SNA, baroreceptor stimulation triggers descending pain inhibitory activity, which contributes to the inverse relationship between resting blood pressure and acute pain.
5. Chronic Pain as a Driver of SNS Overactivation
As introduced above, spinal sympathetic reflex arcs result in the direct activation of sympathetic preganglionic neurons within the IML by ascending sensory/nociceptive pathways. This likely represents the most straightforward neural mechanism by which chronic pain can drive SNS overactivation. Within the dorsal horn, sensory information is directed to: (i) interneurons, which terminate within the same spinal segment, (ii) propriospinal neurons, which terminate within a different spinal segment, and (iii) projecting neurons, which terminate within supraspinal structures. Activation of sympathetic preganglionic neurons occurs either via second-order interneurons, second-order propriospinal neurons, or second-order spinothalamic projecting neurons, which have extensive axon collaterals within the thoracic spinal cord [
167]. Additional interneurons (third order) terminating within the intermediolateral cell column of the spinal cord are likely critical for sensory-sympathetic coupling, as second-order neurons (arising from the dorsal horn) rarely make direct connections with cell bodies of sympathetic preganglionic neurons [
168]. Thus, in the setting of chronic pain, SNS overactivation may result directly from excess spinal nociceptive input via a spinal sympathetic reflex arc.
A second mechanism by which chronic pain may increase sympathetic activity is secondary to chronic cognitive or emotional stress. Excessive sympathetic stress responses are associated with numerous human disorders, including post-traumatic stress disorder, cerebral palsy, traumatic brain injury, autism spectrum disorders, bipolar disorder, epilepsy, and Charcot–Marie–Tooth disease, among others [
169,
170,
171,
172], and can manifest with acute myocardial injury and/or increased susceptibility to sudden cardiac death [
173,
174,
175]. Cardiovascular consequences of stress are similar in all mammals [
67,
176,
177], and chronic or repetitive stress leading to increases in sympathetic activity is one proven cause of hypertension and heart failure [
176,
178]. Pain, especially when present in a chronic form, is a source of discomfort and substantial emotional suffering. In chronic orofacial pain patients, sympathetic responses to mental stressors are significantly greater than responses in pain-free, age and sex-matched controls [
179].
6. Maladaptive Changes in the Neural Circuitry Leading to Sympathetic Overactivation and Chronic Pain
Our understanding of neuroplastic changes associated with chronic pain has been largely made possible by advancements in in vivo imaging using magnetic resonance imaging (MRI) techniques. Enhanced spatial resolution and contrast enable detailed structural morphology studies to be conducted in patients with chronic pain. Functional magnetic resonance imaging (fMRI) studies in humans have demonstrated the coordinated activation of several brain areas in response to noxious somatic and visceral stimuli, including the thalamus, anterior cingulate cortex, insular cortex, primary and secondary sensory cortices, prefrontal cortex, basal ganglia, cerebellum, and amygdala. This network of brain regions involved in both sensory discriminative and emotional-affective aspects of pain is termed the “pain matrix” [
182].
Chronic pain is associated with plastic changes at every level of the neural axis including primary sensory endings, as well as spinal and supraspinal sites [
185]. The changes within the supraspinal centers are particularly important as they may contribute to altered nociceptive processing and perception of pain. Given the anatomical and functional overlap between the central nociceptive and central autonomic networks, the aberrant changes that occur within the supraspinal centers may simultaneously manifest with dysregulated sympathetic control and chronic pain. Recent studies have found that chronic pain sufferers irrespective of the origin of pain, exhibit common brain signatures associated with the loss of gray matter in cortical and subcortical structures such as the prefrontal and insular cortex, orbitofrontal cortex, and pons [
185]. Using connectivity analyses of resting state fMRI, chronic migraine sufferers exhibited reduced gray matter volume and reduced cortical thickness in brain regions involved in the affective processing of pain including dlPFC [
186].
This entry is adapted from the peer-reviewed paper 10.3390/ijms24065378