Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Arif Mahmood | -- | 1974 | 2023-04-11 12:55:59 | | | |
| 2 | Lindsay Dong | Meta information modification | 1974 | 2023-04-13 08:36:49 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Chen, K.; Wang, H.; Ilyas, I.; Mahmood, A.; Hou, L. Microglia and Astrocytes Dysfunction in Parkinson’s Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/42942 (accessed on 23 July 2026).
Chen K, Wang H, Ilyas I, Mahmood A, Hou L. Microglia and Astrocytes Dysfunction in Parkinson’s Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/42942. Accessed July 23, 2026.
Chen, Kun, Haoyang Wang, Iqra Ilyas, Arif Mahmood, Lijun Hou. "Microglia and Astrocytes Dysfunction in Parkinson’s Disease" Encyclopedia, https://encyclopedia.pub/entry/42942 (accessed July 23, 2026).
Chen, K., Wang, H., Ilyas, I., Mahmood, A., & Hou, L. (2023, April 11). Microglia and Astrocytes Dysfunction in Parkinson’s Disease. In Encyclopedia. https://encyclopedia.pub/entry/42942
Chen, Kun, et al. "Microglia and Astrocytes Dysfunction in Parkinson’s Disease." Encyclopedia. Web. 11 April, 2023.
Copy Citation
Parkinson’s disease (PD) is the second most common neurodegenerative disease, with symptoms such as tremor, bradykinesia with rigidity, and depression appearing in the late stage of life. The key hallmark of PD is the loss or death of dopaminergic neurons in the region substantia nigra pars compacta. Neuroinflammation plays a key role in the etiology of PD, and the contribution of immunity-related events spurred the researchers to identify anti-inflammatory agents for the treatment of PD. Microglia and astrocyte dysregulation has been reported in PD. Patients with PD develop neural toxicity, inflammation, and inclusion bodies due to activated microglia and a-synuclein–induced astrocyte conversion into A1 astrocytes.
Parkinson’s disease
astrocytes
microglia
biomarkers
1. Introduction
Among neurodegenerative diseases, Parkinson’s disease (PD) is the second most common after Alzheimer’s disease, affecting 2% of people worldwide. In the United States, Europe, and surrounding countries, the prevalence of PD is high, including in some Asian countries [1]. The progressive degeneration of dopaminergic neurons that occur in the substantia nigra pars compacta (SNpc) causes motor impairment in PD. The mechanism of this neurodegeneration is still largely unknown, but many genetic and environmental factors have been associated with disease occurrence and pathogenesis [2]. Genome-wide association studies (GWAS) suggest that the dysregulation of innate and adaptive immunities may be a key contributor in PD pathogenesis [3]. Several pathoetiological mechanisms contribute to neurodegeneration, including sustained inflammatory processes [4]. The progression of neurodegenerative disorders is typically associated with chronic inflammation as opposed to acute inflammation, which is usually associated with repairing the brain after exposure to a variety of environmental insults such as viral infection, traumatic injury, and toxins [5]. The association of these inflammatory processes with PD are not well defined; however, polymorphism and genetic variants of genes related to immunity have been reported [6][7].
Several factors including hypoxia/ischemia, peripheral nerve injury, trauma (physical and psychological), toxins, and infection activate glial cells in the central nervous system (CNS) [8][9][10]. Physical and chemical support to neurons is provided by glial cells, which also maintain their environment. Neurons communicate with different glial cell in order to properly perform functions [11]. Among many glial cells, mostly microglia and astrocytes play a phagocytotic role by engulfing synapses and apoptotic cells and releasing toxins. Microglia are the resident macrophages of the brain that represent 5–10% of the whole CNS and are necessary for CNS homeostasis [12]. In the brain, glia cells constitute over 50% of all the cells and are divided into several types, including astrocytes. Over 100 years have passed since the discovery of astrocytes, but not much information is available about their functions in neurological diseases. Astrocytes have key functions in the brain, including an active role in circuit building, synaptic turnover, and ion homeostasis. “Astrocyte reactivity”, “astrocyte activation”, “astrogliosis”, “astrocytosis”, and “reactive gliosis” are terms that describe different morphological, molecular, and functional changes in astrocytes [13]. Recent studies suggest that mitochondria plays an important role in the regulation of astrocyte functions such as transmitophagy, calcium signaling, antioxidant production, glutamate regulation, fatty acid metabolism, and neuro-inflammatory activation, which indicates that mitochondrial dysfunction in astrocytes may affect DA neuron health, consequently leading to the death of these neurons [14][15].
A major pathophysiological factor in PD is neuroinflammation in the SNpc caused by astrocyte reactivity. PD etiology has also been associated with expression of several PD-related genes by neurons and glia cells, suggesting that mutated gene products within microglia and astrocytes might play a role in PD progression [16][17]. Many neurodegenerative disorders, including PD, multiple system atrophy (MSA), dementia, and progressive supranuclear palsy (PSP), share common clinical characteristics, which makes the diagnosis more challenging [18]. Understanding neuroinflammation in context with microglia and astrocytes will provide more insight into the mechanism. The identification of key neuroinflammation-based biomarkers can facilitate early detection of PD.
2. Microglia in PD
Microglia are macrophages residing in the CNS and are the primary immune cells. Microglia make up about 10% of the cellular population in a healthy and nondiseased human brain [19]. In the physiological state, microglia mediate several brain functions, primarily synaptic pruning and remodeling and bidirectional signaling, and are important for neural circuits and brain connectivity [20]. Pathological triggers initiate the migration of microglia to the area of injury, where they act as a double-edged sword to either relieve or aggravate the injury. Interestingly, activation of microglia is related to neurodegeneration, the process underlying PD and many other neurodegenerative diseases.
Recently, microglia in PD have drawn prominent attention because of their role as regulators of immunity, primarily in initiation of neuroinflammation in response to proinflammatory molecules. A recent discovery and breakthrough suggest that both the innate immune system and microglia are important for synaptic lopping which describes their contribution in imparting changes to the neurons that surrounds them [21]. The microglial population undergoes controlled cycles of renewal, which maintains their appropriate overall density and may also modulate the relative proportions of different microglial phenotypes [22]. In this way, microglia play a crucial role in maintaining parenchymal homeostasis by being mobile, dynamic, and vigilant observers [23]. Microglia-derived inflammation plays a role in provoking astrocytes to attain neurotoxic functions or lose neurotrophic or synaptoptrophic functionality [24]. Microglia normally maintain functional nerve cells by synaptic pruning, while dysfunctional microglia during synapse phagocytosis led to synaptic dysfunction and neurodegeneration [20]. Moreover, microglia also perform an important function in the clearing of necrotic and apoptotic cells and, further, abolishing aberrant and toxic protein clumps including alpha-synuclein aggregates and β-amyloid [25][26]. Generally, microglia participate in CNS autophagy using phagocytosis and maintaining homeostasis by eliminating the effect of inflammatory response. Microglial autophagy refers to the transport of damaged toxic organelles and protein aggregates to the lysosome by a cascade of events such as autophagosome formation and protein degradation [27]. The dysregulation of autophagic flux in the autophagy pathway hence affects overall the autophagy pathway and which is the major contributor to the PD.
The neuroinflammation in PD is associated with microgliosis. A 2013 genome-wide association study (GWAS) first showed the involvement of the leukocyte antigen gene (HLA-DRA) in the neuroinflammation, which has specifically high expression in microglia [28]. The variant of microglia-triggering receptors (p.R47H), which is expressed on myeloid cells-2 called TREM-2, is associated with PD [9]. In the inflammatory mechanism of PD, TREM2 can affect microglial activation and autophagy through the mTOR/p38 MAPK pathway and thus affect pathological changes in PD [29], as MAPK1 pathway is highlighted in Figure 1. In addition to this, positron emission tomography (PET) studies found that reactive microglia are detected in toxin-induced and transgenic mouse models with PD [30][31]. These findings suggested that microglial activation is correlated with the PD progression and that the microgliosis induces DA neuron (midbrain dopaminergic neuron) toxicity and death.
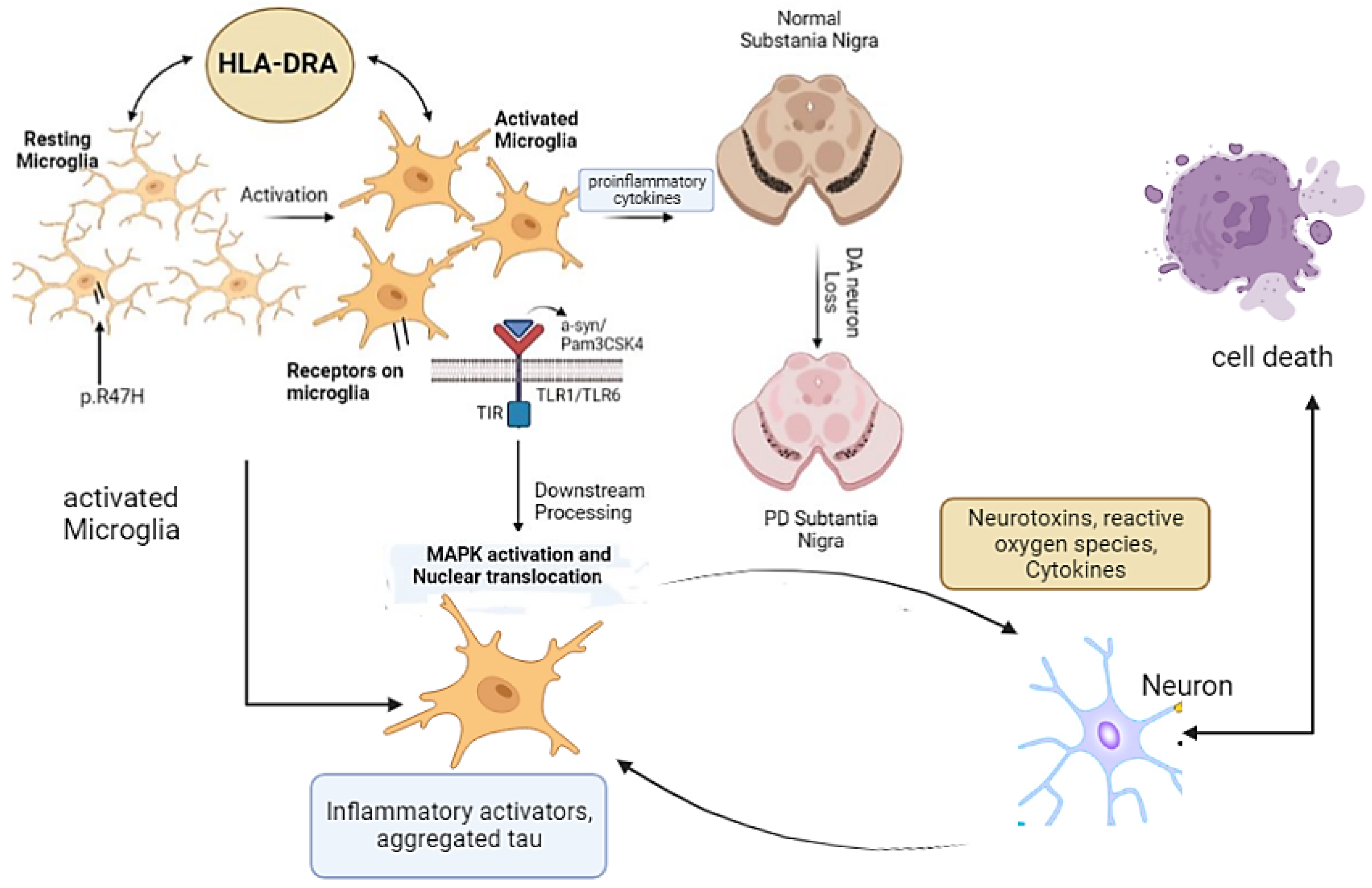
Figure 1. Microglia and microglial receptors in PD. Resting microglia are activated by different stimuli, which ultimately activate MAPK and cell death. Activation of microglia alternatively leads to neuroinflammation and loss of DA neurons and causes PD.
Microglia receptors in the CNS are mostly pattern-recognition receptors (PRRs) that respond to pathogen-associated molecular patterns (PAMPs), one of the prominent types in toll-like receptors (TLRs). Studies showed that microglial receptor TLR1/6 and its downstream pathways enable neuroinflammation by microglia [32][33]. Ligands such as α-Syn and Pam3CSK4 activate TLR2-mediated downstream signaling via the adaptor coreceptor, either TLR1 or TLR6 [24]. Upon α-Syn activation, interaction between the TIR domain activates the kinase activity of the interleukin-1 receptor-associated kinase (IRAK) complex, which then interacts with and activates the TNF receptor-associated factor 6 (TRAF6) via its K63-linked autoubiquitination [22]. This process leads to activation of the transforming growth factor β-activated kinase-1 (TAK1) complex, which then mediates the IκBα degradation. These sequential events lead to the production of proinflammatory cytokines through MAPK activation and the nuclear translocation of NF-κB and p3 [32][34].
The results of the primary microglial culture from the mice treated with the human alpha-synuclein aggregate revealed that alpha-synuclein can directly interact with microglia and can be internalized and transported to autophagosomes. This interaction is mediated by FcγRs, and when the gamma chain is missing, NF-B signaling, and intracellular trafficking are altered. In other studies, using rate primary cultured microglia, the scholars found that alpha-synuclein can induce the expression of matrix metalloproteinases (MMPs) (MMP-1, -3, -8, and -9) [35][36]. PD risk factors are also associated with microglial activation and its progression. The activation of microglia by inducing the expression of matrix metalloproteinases and the subsequent activation of protease-activated receptor-1 are one among the main contributors in PD. Clinical and animal experimental models have suggested a notable higher level of MG during the progression of PD. There was evidence of morphological changes including enlarged and functionally stronger MG than other phagocytoses during proinflammatory response [37].
3. Astrocytes in PD
Astrocytes are specialized glial cells that are present in enormous numbers in the CNS, playing various physiological roles such as synaptic transmission regulation, secretion of neurotrophic molecules, and control of the permeability of the blood–brain barrier (BBB) [38]. It has been observed in PD that there is a disruption of the BBB with differentiation of dopaminergic neurons, which suggests that the normal function of astrocytes is lost with the subsequent progression of PD [39]. Reactive astrocytes are formed in response to injuries in the CNS by releasing a variety of chemokines and cytokines that include tumor necrosis factor alpha (TNF-α) and interleukin-1 beta (IL1β), the release of which aids in PD pathogenesis by toxic gain of function [40]. Another study showed that activation by microglia is induced by pathological α-synuclein, followed by induction of neurotoxic reactive astrocyte by secreting interleukin-1α (IL-1α), TNF-α. Moreover, it was observed in a mouse model of sporadic PD that pathological α-synuclein prevents α-synuclein-induced microglial activation and protects the astrocyte against dopaminergic degeneration [41], as illustrated in Figure 2.
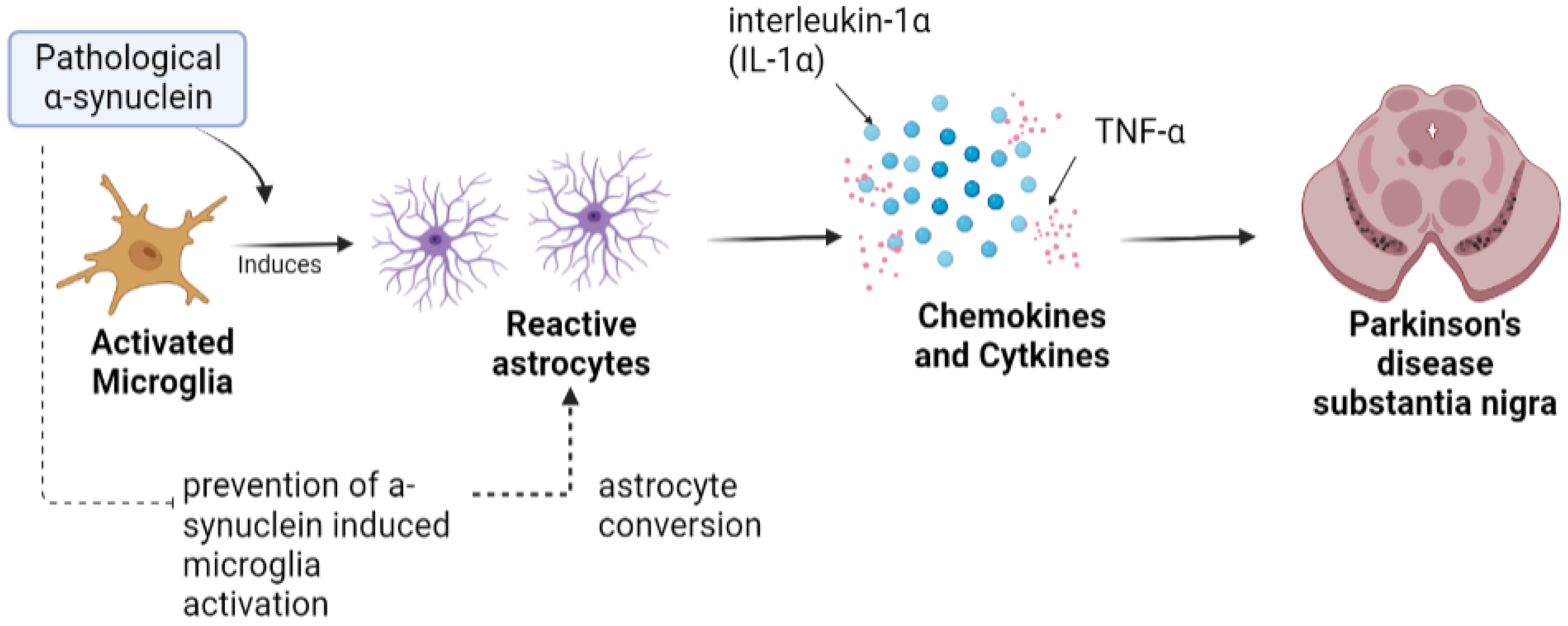
Figure 2. Astrocytes in Parkinson’s disease. Toxic alpha-synuclein convert resting astrocytes into reactive astrocyte, which further disrupts balance of cytokines that can possibly result in loss of DA neurons.
The identification of PD progression in pathological contexts leads to the accumulation of a-synuclein-positive cytoplasmic inclusions in neurons. Alpha-synuclein is encoded by the SNCA gene, and mutation or disruption in this gene leads to PD. It has been observed that there is low expression of SNCA in astrocytes compared to its expression in neurons [42]. The protein α-synuclein plays a physiological role such as mediation in astrocytes. Its deficiency disrupts astrocyte fatty acid uptake and trafficking. Pathologic α-synuclein accumulated is evident in the postmortem brains of PD patients [43]. It has been also found that cell-to-cell transmission of α-synuclein contributes to the predominant expression of alpha-synuclein in neurons, which leads to formation of inclusion bodies in astrocytes [44][45]. The astrocytes that are accumulated with α-synuclein have shown proinflammatory cytokines such as IL-1 and IL-6 as well as some chemokines [45]. These chemokines contribute to the dysfunction of astrocytes, leading to PD progression and pathogenesis. The TLR-4 independent endocytosis pathway showed that, astrocytes can take up alpha-synuclein [46][47]. The endocytosed alpha-synuclein is localized to the lysosome, suggesting astrocyte’s role in alpha-synuclein’s degradation and removal. A high concentration of alpha-synuclein extracellularly has been shown to induce TLR4-dependent inflammatory response in primary astrocyte cultures [40]. In other words, this response is concentration-dependent; when the concentration of alpha-synuclein exceeds the threshold, then an inflammatory response is induced, and PD begins to develop, whereas in other cases when alpha-synuclein secretes from the neuron, astrocytes endocytose and degrade them [48].
Astrocyte dysfunction is possible due to five main mechanisms [49]: first, through the aquaporin-4 (AQP4) water channels, which are mislocalized and distant from the astrocytes, and second, through reduction in the neuroprotective capacity of astrocytes. The aggregation of alpha-synuclein leads to a decrease in the neurotrophic factor release. This damages the astrocyte function and results in decreased neuroprotection of astrocytes. In the third mechanism, astrocyte contributes to neural toxicity through inflammatory signaling via an increase in inflammasome pathways of TLR4, IFN-g, and NLPR3. In the fourth mechanism, impaired astrocyte proliferation occurs, which reduces the capacity of cells to respond. In the fifth mechanism, the reduction in glutamate uptake leads to an increase in extracellular glutamate, thus contributing neuronal excitotoxicity [43][50][51].
References
- Mahmood, A.; Shah, A.A.; Umair, M.; Wu, Y.; Khan, A. Recalling the pathology of Parkinson’s disease; lacking exact figure of prevalence and genetic evidence in Asia with an alarming outcome: A time to step-up. Clin. Genet. 2021, 100, 659–677.
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 22, 657–673.
- Pierce, S.; Coetzee, G.A. Parkinson’s disease-associated genetic variation is linked to quantitative expression of inflammatory genes. PLoS ONE 2017, 12, e0175882.
- Walker, K.A. Inflammation and neurodegeneration: Chronicity matters. Aging 2019, 11, 3.
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42.
- Wang, J.; Liu, Y.; Liu, Y.; Zhu, K.; Xie, A. The association between TLR3 rs3775290 polymorphism and sporadic Parkinson’s disease in Chinese Han population. Neurosci. Lett. 2020, 728, 135005.
- Ulhaq, Z.S.; Garcia, C.P. Inflammation-related gene polymorphisms associated with Parkinson’s disease: An updated meta-analysis. Egypt. J. Med. Hum. Genet. 2020, 21, 14.
- Graeber, M.B.; Streit, W.J. Microglia: Biology and pathology. Acta Neuropathol. 2010, 119, 89–105.
- Graeber, M.B. Changing face of microglia. Science 2010, 330, 783–788.
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 2009, 29, 3974–3980.
- Pannese, E. Neurocytology: Fine Structure of Neurons, Nerve Processes, and Neuroglial Cells; Springer: Berlin/Heidelberg, Germany, 2015.
- Holtbernd, F.; Ma, Y.; Peng, S.; Schwartz, F.; Timmermann, L.; Kracht, L.; Fink, G.R.; Tang, C.C.; Eidelberg, D.; Eggers, C. Dopaminergic correlates of metabolic network activity in Parkinson’s disease. Hum. Brain Mapp. 2015, 36, 3575–3585.
- Escartin, C.; Guillemaud, O.; Carrillo-de Sauvage, M. Questions and (some) answers on reactive astrocytes. Glia 2019, 67, 2221–2247.
- Sher, A.A.; Gao, A.; Coombs, K.M. Autophagy Modulators Profoundly Alter the Astrocyte Cellular Proteome. Cells 2020, 9, 805.
- Pang, S.Y.-Y.; Ho, P.W.-L.; Liu, H.-F.; Leung, C.-T.; Li, L.; Chang, E.E.S.; Ramsden, D.B.; Ho, S.-L. The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl. Neurodegener. 2019, 8, 23.
- Booth, F.W.; Roberts, C.K.; Thyfault, J.P.; Ruegsegger, G.N.; Toedebusch, R.G. Role of inactivity in chronic diseases: Evolutionary insight and pathophysiological mechanisms. Physiol. Rev. 2017, 97, 1351–1402.
- Wang, J.-Y.; Zhuang, Q.-Q.; Zhu, L.-B.; Zhu, H.; Li, T.; Li, R.; Chen, S.-F.; Huang, C.-P.; Zhang, X.; Zhu, J.-H. Meta-analysis of brain iron levels of Parkinson’s disease patients determined by postmortem and MRI measurements. Sci. Rep. 2016, 6, 36669.
- Meara, J.; Bhowmick, B.K.; Hobson, P. Accuracy of diagnosis in patients with presumed Parkinson’s disease. Age Ageing 1999, 28, 99–102.
- Mittelbronn, M.; Dietz, K.; Schluesener, H.J.; Meyermann, R. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 2001, 101, 249–255.
- Paolicelli, R.C.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; Ragozzino, D.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458.
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361.
- Askew, K.; Olmos-Alonso, A.; Garcia-Moreno, F.; Liang, Y.; Richardson, P.; Tipton, T.; Chapman, M.A.; Riecken, K.; Beccari, S.; Sierra, A.; et al. Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep. 2017, 18, 391–405.
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318.
- Lev, N.; Barhum, Y.; Ben-Zur, T.; Melamed, E.; Steiner, I.; Offen, D. Knocking out DJ-1 attenuates astrocytes neuroprotection against 6-hydroxydopamine toxicity. J. Mol. Neurosci. 2013, 50, 542–550.
- Spangenberg, E.E.; Lee, R.J.; Najafi, A.R.; Rice, R.A.; Elmore, M.R.P.; Blurton-Jones, M.; West, B.L.; Green, K.N. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain 2016, 139, 1265–1281.
- Yuan, J.; Liu, H.; Zhang, H.; Wang, T.; Zheng, Q.; Li, Z. Controlled activation of TRPV1 channels on microglia to boost their autophagy for clearance of alpha-Synuclein and enhance therapy of Parkinson’s disease. Adv. Mater. 2022, 34, e2108435.
- Hu, Y.; Reggiori, F. Molecular regulation of autophagosome formation. Biochem. Soc. Trans. 2022, 50, 55–69.
- Ronan, J.L.; Wu, W.; Crabtree, G.R. From neural development to cognition: Unexpected roles for chromatin. Nat. Rev. Genet. 2013, 14, 347–359.
- Huang, W.; Lv, Q.; Xiao, Y.; Zhong, Z.; Hu, B.; Yan, S.; Yan, Y.; Zhang, J.; Shi, T.; Jiang, L.; et al. Triggering Receptor Expressed on Myeloid Cells 2 Protects Dopaminergic Neurons by Promoting Autophagy in the Inflammatory Pathogenesis of Parkinson’s Disease. Front. Neurosci. 2021, 15, 745815.
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with (R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412.
- Bartels, A.; Willemsen, A.T.M.; Doorduin, J.; de Vries, E.F.J.; Dierckx, R.A.; Leenders, K.L. -PK11195 PET: Quantification of neuroinflammation and a monitor of anti-inflammatory treatment in Parkinson’s disease? Park. Relat. Disord. 2010, 16, 57–59.
- Kim, C.C.; Nakamura, M.C.; Hsieh, C.L. Brain trauma elicits non-canonical macrophage activation states. J. Neuroinflamm. 2016, 13, 117.
- Hanisch, U.-K. Functional diversity of microglia–how heterogeneous are they to begin with? Front. Cell. Neurosci. 2013, 7, 65.
- Hoenen, C.; Gustin, A.; Birck, C.; Kirchmeyer, M.; Beaume, N.; Felten, P.; Grandbarbe, L.; Heuschling, P.; Heurtaux, T. Alpha-synuclein proteins promote pro-inflammatory cascades in microglia: Stronger effects of the A53T mutant. PLoS ONE 2016, 11, e0162717.
- Cao, S.; Standaert, D.G.; Harms, A.S. The gamma chain subunit of Fc receptors is required for alpha-synuclein-induced pro-inflammatory signaling in microglia. J. Neuroinflamm. 2012, 9, 259.
- Lee, E.-J.; Woo, M.-S.; Moon, P.-G.; Baek, M.-C.; Choi, I.-Y.; Kim, W.-K.; Junn, E.; Kim, H.-S. α-Synuclein activates microglia by inducing the expressions of matrix metalloproteinases and the subsequent activation of protease-activated receptor-1. J. Immunol. 2010, 185, 615–623.
- Li, S.; Bi, G.; Han, S.; Huang, R. MicroRNAs Play a Role in Parkinson’s Disease by Regulating Microglia Function: From Pathogenetic Involvement to Therapeutic Potential. Front. Mol. Neurosci. 2022, 14, 744942.
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Astrocyte–neuron metabolic relationships: For better and for worse. Trends Neurosci. 2011, 34, 76–87.
- Cabezas, R.; Ãvila, M.; Gonzalez, J.; El-Bachá, R.S.; Báez, E.; García-Segura, L.M.; Coronel, J.C.J.; Capani, F.; Cardona-Gomez, G.P.; Barreto, G.E. Astrocytic modulation of blood brain barrier: Perspectives on Parkinson’s disease. Front. Cell. Neurosci. 2014, 8, 211.
- Choi, S.S.; Lee, H.J.; Lim, I.; Satoh, J.; Kim, S.U. Human astrocytes: Secretome profiles of cytokines and chemokines. PLoS ONE 2014, 9, e92325.
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487.
- Solano, S.M.; Miller, D.W.; Augood, S.J.; Young, A.B.; Penney, J.B. Expression of α-synuclein, parkin, and ubiquitin carboxy-terminal hydrolase L1 mRNA in human brain: Genes associated with familial Parkinson’s disease. Ann. Neurol. 2000, 47, 201–210.
- Braak, H.; Sastre, M.; Del Tredici, K. Development of α-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241.
- Hansen, C.; Angot, E.; Bergström, A.-L.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K.; et al. α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Investig. 2011, 121, 715–725.
- Lee, H.-J.; Kim, C.; Lee, S.-J. Alpha-synuclein stimulation of astrocytes: Potential role for neuroinflammation and neuroprotection. Oxidative Med. Cell. Longev. 2010, 3, 283–287.
- Rannikko, E.H.; Weber, S.S.; Kahle, P.J. Exogenous α-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci. 2015, 16, 57.
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360.
- Klegeris, A.; Giasson, B.I.; Zhang, H.; Maguire, J.; Pelech, S.; McGeer, P.L. Alpha-synuclein and its disease-causing mutants induce ICAM-1 and IL-6 in human astrocytes and astrocytoma cells. FASEB J. 2006, 20, 2000–2008.
- Gu, X.-L.; Long, C.-X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic expression of Parkinson’s disease-related A53T α-synuclein causes neurodegeneration in mice. Mol. Brain 2010, 3, 12.
- Kim, J.-M.; Cha, S.-H.; Choi, Y.R.; Jou, I.; Joe, E.-H.; Park, S.M. DJ-1 deficiency impairs glutamate uptake into astrocytes via the regulation of flotillin-1 and caveolin-1 expression. Sci. Rep. 2016, 6, 28823.
- Mullett, S.J.; Hinkle, D.A. DJ-1 knock-down in astrocytes impairs astrocyte-mediated neuroprotection against rotenone. Neurobiol. Dis. 2009, 33, 28–36.
More
Information
Subjects:
Immunology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.2K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
13 Apr 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No