Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Arif Mahmood and Version 2 by Lindsay Dong.
Parkinson’s disease (PD) is the second most common neurodegenerative disease, with symptoms such as tremor, bradykinesia with rigidity, and depression appearing in the late stage of life. The key hallmark of PD is the loss or death of dopaminergic neurons in the region substantia nigra pars compacta. Neuroinflammation plays a key role in the etiology of PD, and the contribution of immunity-related events spurred the researchers to identify anti-inflammatory agents for the treatment of PD. Microglia and astrocyte dysregulation has been reported in PD. Patients with PD develop neural toxicity, inflammation, and inclusion bodies due to activated microglia and a-synuclein–induced astrocyte conversion into A1 astrocytes.
- Parkinson’s disease
- astrocytes
- microglia
- biomarkers
1. Introduction
Among neurodegenerative diseases, Parkinson’s disease (PD) is the second most common after Alzheimer’s disease, affecting 2% of people worldwide. In the United States, Europe, and surrounding countries, the prevalence of PD is high, including in some Asian countries [1]. The progressive degeneration of dopaminergic neurons that occur in the substantia nigra pars compacta (SNpc) causes motor impairment in PD. The mechanism of this neurodegeneration is still largely unknown, but many genetic and environmental factors have been associated with disease occurrence and pathogenesis [2]. Genome-wide association studies (GWAS) suggest that the dysregulation of innate and adaptive immunities may be a key contributor in PD pathogenesis [3]. Several pathoetiological mechanisms contribute to neurodegeneration, including sustained inflammatory processes [4]. The progression of neurodegenerative disorders is typically associated with chronic inflammation as opposed to acute inflammation, which is usually associated with repairing the brain after exposure to a variety of environmental insults such as viral infection, traumatic injury, and toxins [5]. The association of these inflammatory processes with PD are not well defined; however, polymorphism and genetic variants of genes related to immunity have been reported [6][7][6,7].
Several factors including hypoxia/ischemia, peripheral nerve injury, trauma (physical and psychological), toxins, and infection activate glial cells in the central nervous system (CNS) [8][9][10][9,10,11]. Physical and chemical support to neurons is provided by glial cells, which also maintain their environment. Neurons communicate with different glial cell in order to properly perform functions [11][12]. Among many glial cells, mostly microglia and astrocytes play a phagocytotic role by engulfing synapses and apoptotic cells and releasing toxins. Microglia are the resident macrophages of the brain that represent 5–10% of the whole CNS and are necessary for CNS homeostasis [12][13]. In the brain, glia cells constitute over 50% of all the cells and are divided into several types, including astrocytes. Over 100 years have passed since the discovery of astrocytes, but not much information is available about their functions in neurological diseases. Astrocytes have key functions in the brain, including an active role in circuit building, synaptic turnover, and ion homeostasis. “Astrocyte reactivity”, “astrocyte activation”, “astrogliosis”, “astrocytosis”, and “reactive gliosis” are terms that describe different morphological, molecular, and functional changes in astrocytes [13][14]. Recent studies suggest that mitochondria plays an important role in the regulation of astrocyte functions such as transmitophagy, calcium signaling, antioxidant production, glutamate regulation, fatty acid metabolism, and neuro-inflammatory activation, which indicates that mitochondrial dysfunction in astrocytes may affect DA neuron health, consequently leading to the death of these neurons [14][15][15,16].
A major pathophysiological factor in PD is neuroinflammation in the SNpc caused by astrocyte reactivity. PD etiology has also been associated with expression of several PD-related genes by neurons and glia cells, suggesting that mutated gene products within microglia and astrocytes might play a role in PD progression [16][17][19,20]. Many neurodegenerative disorders, including PD, multiple system atrophy (MSA), dementia, and progressive supranuclear palsy (PSP), share common clinical characteristics, which makes the diagnosis more challenging [18][21]. Understanding neuroinflammation in context with microglia and astrocytes will provide more insight into the mechanism. The identification of key neuroinflammation-based biomarkers can facilitate early detection of PD.
2. Microglia in PD
Microglia are macrophages residing in the CNS and are the primary immune cells. Microglia make up about 10% of the cellular population in a healthy and nondiseased human brain [19][30]. In the physiological state, microglia mediate several brain functions, primarily synaptic pruning and remodeling and bidirectional signaling, and are important for neural circuits and brain connectivity [20][31]. Pathological triggers initiate the migration of microglia to the area of injury, where they act as a double-edged sword to either relieve or aggravate the injury. Interestingly, activation of microglia is related to neurodegeneration, the process underlying PD and many other neurodegenerative diseases. Recently, microglia in PD have drawn prominent attention because of their role as regulators of immunity, primarily in initiation of neuroinflammation in response to proinflammatory molecules. A recent discovery and breakthrough suggest that both the innate immune system and microglia are important for synaptic lopping which describes their contribution in imparting changes to the neurons that surrounds them [21][32]. The microglial population undergoes controlled cycles of renewal, which maintains their appropriate overall density and may also modulate the relative proportions of different microglial phenotypes [22][33]. In this way, microglia play a crucial role in maintaining parenchymal homeostasis by being mobile, dynamic, and vigilant observers [23][34]. Microglia-derived inflammation plays a role in provoking astrocytes to attain neurotoxic functions or lose neurotrophic or synaptoptrophic functionality [24][35]. Microglia normally maintain functional nerve cells by synaptic pruning, while dysfunctional microglia during synapse phagocytosis led to synaptic dysfunction and neurodegeneration [20][31]. Moreover, microglia also perform an important function in the clearing of necrotic and apoptotic cells and, further, abolishing aberrant and toxic protein clumps including alpha-synuclein aggregates and β-amyloid [25][26][36,37]. Generally, microglia participate in CNS autophagy using phagocytosis and maintaining homeostasis by eliminating the effect of inflammatory response. Microglial autophagy refers to the transport of damaged toxic organelles and protein aggregates to the lysosome by a cascade of events such as autophagosome formation and protein degradation [27][38]. The dysregulation of autophagic flux in the autophagy pathway hence affects overall the autophagy pathway and which is the major contributor to the PD. The neuroinflammation in PD is associated with microgliosis. A 2013 genome-wide association study (GWAS) first showed the involvement of the leukocyte antigen gene (HLA-DRA) in the neuroinflammation, which has specifically high expression in microglia [28][39]. The variant of microglia-triggering receptors (p.R47H), which is expressed on myeloid cells-2 called TREM-2, is associated with PD [9][10]. In the inflammatory mechanism of PD, TREM2 can affect microglial activation and autophagy through the mTOR/p38 MAPK pathway and thus affect pathological changes in PD [29][40], as MAPK1 pathway is highlighted in Figure 1. In addition to this, positron emission tomography (PET) studies found that reactive microglia are detected in toxin-induced and transgenic mouse models with PD [30][31][41,42]. These findings suggested that microglial activation is correlated with the PD progression and that the microgliosis induces DA neuron (midbrain dopaminergic neuron) toxicity and death.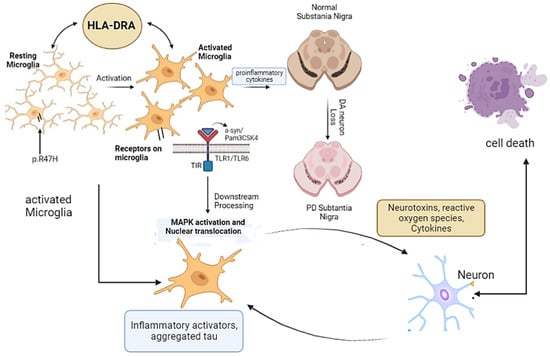
Figure 1. Microglia and microglial receptors in PD. Resting microglia are activated by different stimuli, which ultimately activate MAPK and cell death. Activation of microglia alternatively leads to neuroinflammation and loss of DA neurons and causes PD.
3. Astrocytes in PD
Astrocytes are specialized glial cells that are present in enormous numbers in the CNS, playing various physiological roles such as synaptic transmission regulation, secretion of neurotrophic molecules, and control of the permeability of the blood–brain barrier (BBB) [38][49]. It has been observed in PD that there is a disruption of the BBB with differentiation of dopaminergic neurons, which suggests that the normal function of astrocytes is lost with the subsequent progression of PD [39][50]. Reactive astrocytes are formed in response to injuries in the CNS by releasing a variety of chemokines and cytokines that include tumor necrosis factor alpha (TNF-α) and interleukin-1 beta (IL1β), the release of which aids in PD pathogenesis by toxic gain of function [40][51]. Another study showed that activation by microglia is induced by pathological α-synuclein, followed by induction of neurotoxic reactive astrocyte by secreting interleukin-1α (IL-1α), TNF-α. Moreover, it was observed in a mouse model of sporadic PD that pathological α-synuclein prevents α-synuclein-induced microglial activation and protects the astrocyte against dopaminergic degeneration [41][52], as illustrated in Figure 2.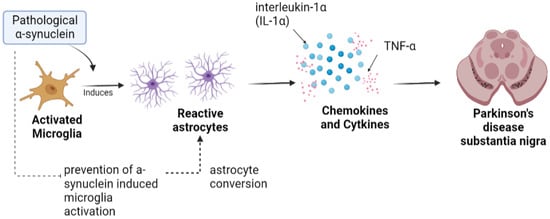
Figure 2. Astrocytes in Parkinson’s disease. Toxic alpha-synuclein convert resting astrocytes into reactive astrocyte, which further disrupts balance of cytokines that can possibly result in loss of DA neurons.