 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Zijun Y. Xu-Monette | -- | 3620 | 2023-04-06 16:32:08 | | | |
| 2 | Jessie Wu | Meta information modification | 3620 | 2023-04-07 05:20:48 | | | | |
| 3 | Jessie Wu | Meta information modification | 3620 | 2023-04-07 05:22:43 | | |
Video Upload Options
Lymphoma is a heterogeneous group of diseases that often require their metabolism programs to fulfill the demand of cell proliferation. Features of metabolism in lymphoma cells include high glucose uptake, deregulated expression of enzymes related to glycolysis, dual capacity for glycolytic and oxidative metabolism, elevated glutamine metabolism, and fatty acid synthesis. These aberrant metabolic changes lead to tumorigenesis, disease progression, and resistance to lymphoma chemotherapy. Metabolic reprogramming, including glucose, nucleic acid, fatty acid, and amino acid metabolism, is a dynamic process caused not only by genetic and epigenetic changes, but also by changes in the microenvironment affected by viral infections. Notably, some critical metabolic enzymes and metabolites may play vital roles in lymphomagenesis and progression.
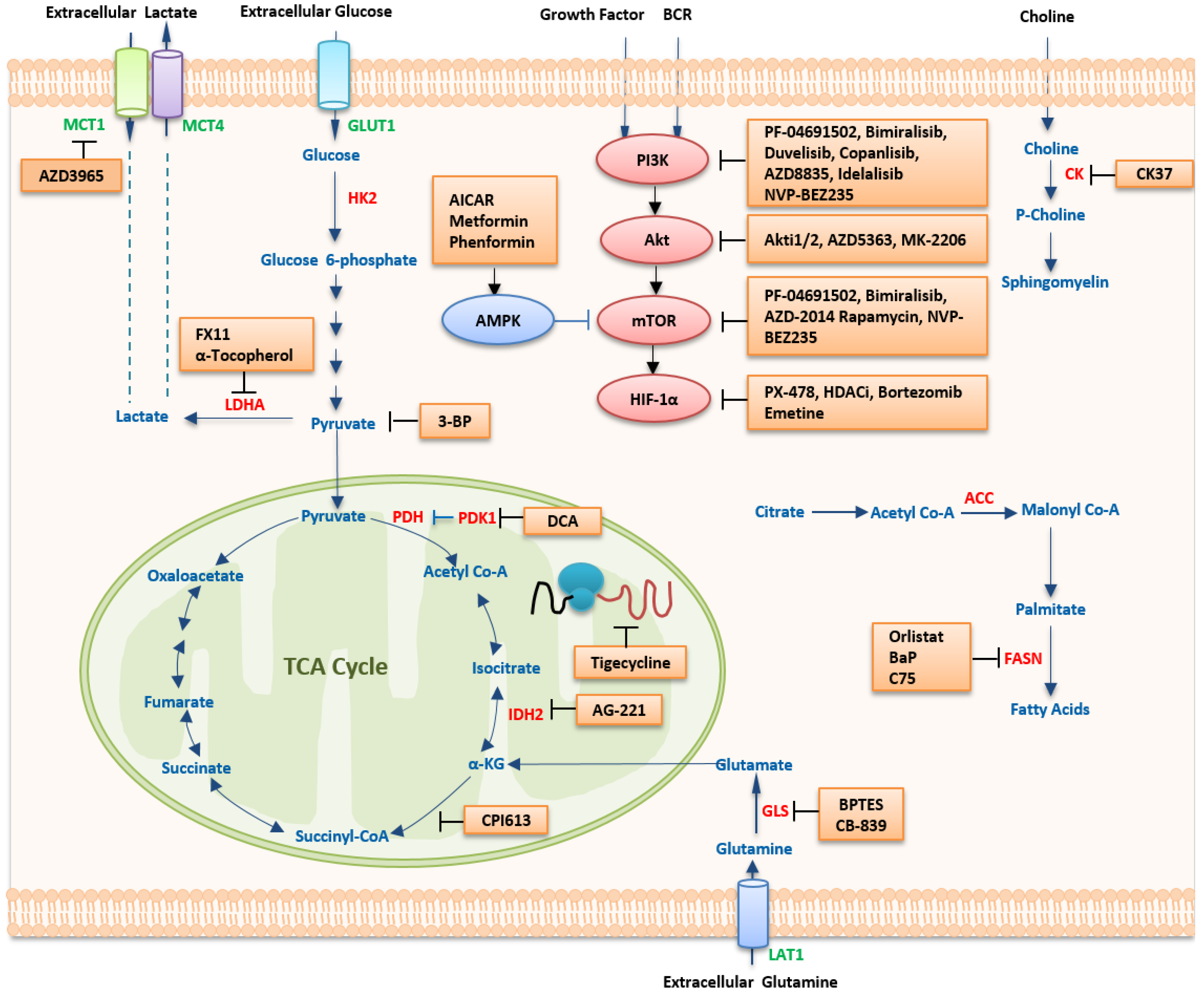
1. Therapeutic Strategies Targeting Metabolism
| Agent | Target | Mechanism | Type of Diseases |
|---|---|---|---|
| methotrexate | DHFR | folate to THF conversion | prophylaxis and treatment of CNS lymphoma |
| IM156 | mitochondrial complex I inhibitor | mitochondrial oxidative phosphorylation and NADH oxidation | lymphomas (NCT03272256) |
| IACS-010759 | oxidative phosphorylation inhibitor | mitochondrial oxidative phosphorylation and NADH oxidation | R/R AML (NCT02882321) |
| AZD-3965 | MCT1 | mediate the bidirectional transport of lactatein and out of cells | DLBCL, BL (NCT01791595) |
| CPI-613 | mitochondria | oxidative metabolism | T-cell NHL (NCT04217317), R/R BL or HGBCL (NCT03793140) |
| IM156 | mitochondria | complex I | advanced lymphoma (NCT03272256) |
| IACS-010759 | mitochondria | complex I | R/R AML (NCT02882321) |
| ibrutinib | BTK inhibitor | downstream pro-proliferative kinase of BCR signal | MCL, CLL/SLL, WM, MZL, cGVHD (NCT02169180, NCT04771507, NCT02604511, NCT04212013, NCT05348096) |
| acalabrutinib | MCL, CLL/SLL, MZL (NCT02213926, NCT04505254, NCT04646395) | ||
| rapalogs | mTORC1 inhibitor | rapamycin analogues | MCL |
| temsirolimus | mTORC1 inhibitor | cell cycle arrest in the G1 phase, inhibits tumor angiogenesis by reducing synthesis of VEGF | MCL (NCT01078142, NCT01180049), HL (NCT01902160), R/R NHL (NCT01281917) |
| idelalisib | PI3Kδ inhibitor | against PI3Kδ isoforms | iNHL (NCT01282424), HL (NCT01393106), FL (NCT03568929), CLL (NCT03582098) |
| copanlisib | PI3Kδ and PI3Kα inhibitor | against PI3K-α and PI3K-δ isoforms | iNHL, DLBCL, MCL, PTCL (NCT05217914, NCT04433182, NCT04939272, NCT03877055, NCT03052933) |
| duvelisib | PI3Kδ and PI3Kγinhibitor | against PI3Kδ and PI3Kγ isoforms | CLL/SLL, DLBCL, PTCL (NCT02004522, NCT04890236, NCT04803201) |
| umbralisib | PI3Kδ and casein kinase-1 epsilon inhibitor | against PI3Kδisoforms and casein kinase-1 epsilon | MZL, FL, CLL, WM (NCT03919175, NCT03364231, NCT02535286) |
| L-asparaginase | asparagine | inhibit protein biosynthesis in lymphoblasts | ALL (NCT01518517, NCT00506597), NK/T cell lymphoma (NCT00854425) |
| Telaglenastat (CB-839) | glutaminase inhibitor | glutamine to glutamate conversion | NHL (NCT02071888), including MCL, WM, TCL |
| AG-270 | MAT2A | production of S-adenosylmethionine | advanced lymphoma (NCT03361358, NCT03435250) |
| Devimistat (CPI-613) | lipoate analog | mitochondrial oxidative metabolism | R/R T-cell NHL (NCT04217317) |
1.1. Targeting Metabolic Pathways
1.1.1. Targeting Glucose Metabolism
| Pathway | Compound | Application | Development Stage | Ref |
|---|---|---|---|---|
| HK2 | 2-DG | B-NHL | cell lines | [2] |
| 3-BrPA | BL | cell lines, mice | [6][7] | |
| Pyruvate | 3-BP | T-NHL | mice (DL) | [8] |
| LDHA | FX11 | BL | cell line | [9][10] |
| α-Tocopherol | T-NHL | mice (DL) | [10] | |
| PDK | DCA | DL | mice | [13][14] |
| Mitochondrial protein translation | Tigecycline | OxPhos-DLBCL cell lines | FDA-approved | [16] |
| Glutaminase | BPTES | BL | mice | [20] |
| Glutamine uptake | L-asparaginase | NHL | cell lines | [21] |
| SHMT1/2 | SHIN1 | DLBCL | cell lines | [22] |
| FASN | orlistat | MCL | cell lines | [23][24] |
| T-NHL | cell lines, mice | [25] | ||
| C75 | DLBCL, PEL, and B-NHL | cell lines | [26][27] | |
| NA | BaP | BL | patients | [28] |
| PPARα | Fenofibrate | B-NHL | mice | [29] |
| Choline kinase | CK37 | T-NHL | mice | [30] |
| HIF-1α | PX-478 | PEL | cell lines | [31] |
| PCI-24781(HDACi) | DLBCL | Phase 1/2 | [32] | |
| SAHA(HDACi) | B-NHL | cell lines, mice | [33] | |
| MYC | 10058-F4 | DLBCL | cell lines | [34][35][36] |
| PI3K | LY294002 | B-NHL | cell lines | [37] |
| AZD8835 | ABC-DLBCL | cell lines | [38] | |
| AKT | Akti1/2 | PEL | cell lines | [39] |
| AZD-5363 | PTEN-deficient DLBCL | cell lines, mice | [38] | |
| MK-2206 | ABC-DLBCL | mice | [40] | |
| NaB (HDACi) | BL | cell lines | [41] | |
| mTOR | Rapamycin | ALCL, NHL | cell lines | [42][43] |
| mTOR C1/2 | AZD-2014 | MCL | cell lines | [44] |
| Dual inhibitor of PI3K and mTOR | NVP-BEZ235 | PEL | mice | [45] |
| PF-04091502 | PEL | cell lines | [39] | |
| Bimiralisib (PQR309) | DLBCL, MCL, SMZL, CLL, HL, and ALCL | cell lines, mice | [46] | |
| AMPK | AICAR | MCL, SMZL, FL, and CLL | cell lines, mice and patients | [47][48][49][50][51][52] |
| Metformin | B and T-NHL | cell lines, mice | [53][54] | |
| Phenformin | PTEN-deficient T-cell lymphomas | cell lines | [54] |
1.1.2. Targeting Amino Acid Metabolism
1.1.3. Targeting Lipid Metabolism
1.2. Targeting Oncogenic Regulators
1.2.1. HIF-1α Inhibitors
1.2.2. MYC Inhibitors
1.2.3. Targeting The PI3K/mTOR Pathway
PI3K Inhibitors
Akt Inhibitors
mTOR Inhibitors
Dual Inhibitors of PI3K and mTOR
1.2.4. AMPK Activators
2. Potential Metabolic Biomarkers of Lymphoma
| Subtype | Control | Metabolites | Type of Samples | Origin | Method | Clinical Relevance | Ref |
|---|---|---|---|---|---|---|---|
| DLBCL | healthy people | 2-AG | serum | patients | LC-MS/MS | pathogenesis or progression | [77] |
| BL | Normal mice | glutamate, glycerol, and choline | serum | mice | NMR and MS | diagnosis and prognosis | [78] |
| Refractory or early relapse DLBCL patients | cured patients | ↑lysine, arginine, cadaverine, 2-HB | serum | patients | NMR | high-risk of failing to immunochemotherapy | [79] |
| CTCL | control samples | ↑GLT ↓adenosine monophosphate ↑CTP ↑prostaglandins, pyrimidine, mevalonate pathway ↓tryptophan ↑PRPP |
skin and plasma | mice | UHPLC-QTOF | progress of carcinogenesis, leads to CTCL further development. | [80] |
| MYC-induced lymphomas | normal tissue | ↑glycerophosphoglycerols, cardiolipins and monounsaturated fatty acids | tissue and cells | patients and cell lines | DESI-MSI | MYC regulates cellular metabolism in cancer | [81] |
References
- Geschwind, J.F.; Georgiades, C.S.; Ko, Y.H.; Pedersen, P.L. Recently elucidated energy catabolism pathways provide opportunities for novel treatments in hepatocellular carcinoma. Expert Rev. Anticancer Ther. 2004, 4, 449–457.
- Pang, Y.Y.; Wang, T.; Chen, F.Y.; Wu, Y.L.; Shao, X.; Xiao, F.; Huang, H.H.; Zhong, H.; Zhong, J.H. Glycolytic inhibitor 2-deoxy-d-glucose suppresses cell proliferation and enhances methylprednisolone sensitivity in non-Hodgkin lymphoma cells through down-regulation of HIF-1α and c-MYC. Leuk. Lymphoma 2015, 56, 1821–1830.
- Zagorodna, O.; Martin, S.M.; Rutkowski, D.T.; Kuwana, T.; Spitz, D.R.; Knudson, C.M. 2-deoxyglucose-induced toxicity is regulated by Bcl-2 family members and is enhanced by antagonizing Bcl-2 in lymphoma cell lines. Oncogene 2012, 31, 2738–2749.
- Meynet, O.; Bénéteau, M.; Jacquin, M.A.; Pradelli, L.A.; Cornille, A.; Carles, M.; Ricci, J.E. Glycolysis inhibition targets Mcl-1 to restore sensitivity of lymphoma cells to ABT-737-induced apoptosis. Leukemia 2012, 26, 1145–1147.
- Robinson, G.L.; Dinsdale, D.; Macfarlane, M.; Cain, K. Switching from aerobic glycolysis to oxidative phosphorylation modulates the sensitivity of mantle cell lymphoma cells to TRAIL. Oncogene 2012, 31, 4996–5006.
- Ko, Y.H.; Pedersen, P.L.; Geschwind, J.F. Glucose catabolism in the rabbit VX2 tumor model for liver cancer: Characterization and targeting hexokinase. Cancer Lett. 2001, 173, 83–91.
- Schaefer, N.G.; Geschwind, J.F.; Engles, J.; Buchanan, J.W.; Wahl, R.L. Systemic administration of 3-bromopyruvate in treating disseminated aggressive lymphoma. Transl. Res. 2012, 159, 51–57.
- Yadav, S.; Pandey, S.K.; Kumar, A.; Kujur, P.K.; Singh, R.P.; Singh, S.M. Antitumor and chemosensitizing action of 3-bromopyruvate: Implication of deregulated metabolism. Chem. Biol. Interact. 2017, 270, 73–89.
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042.
- Nilsson, L.M.; Forshell, T.Z.; Rimpi, S.; Kreutzer, C.; Pretsch, W.; Bornkamm, G.W.; Nilsson, J.A. Mouse genetics suggests cell-context dependency for Myc-regulated metabolic enzymes during tumorigenesis. PLoS Genet. 2012, 8, e1002573.
- Curtis, N.J.; Mooney, L.; Hopcroft, L.; Michopoulos, F.; Whalley, N.; Zhong, H.; Murray, C.; Logie, A.; Revill, M.; Byth, K.F.; et al. Pre-clinical pharmacology of AZD3965, a selective inhibitor of MCT1, DLBCL, NHL and Burkitt’s lymphoma anti-tumor activity. Oncotarget 2017, 8, 69219–69236.
- Beloueche-Babari, M.; Wantuch, S.; Casals Galobart, T.; Koniordou, M.; Parkes, H.G.; Arunan, V.; Chung, Y.L.; Eykyn, T.R.; Smith, P.D.; Leach, M.O. MCT1 Inhibitor AZD3965 Increases Mitochondrial Metabolism, Facilitating Combination Therapy and Noninvasive Magnetic Resonance Spectroscopy. Cancer Res. 2017, 77, 5913–5924.
- Schultz, I.R.; Shangraw, R.E. Effect of short-term drinking water exposure to dichloroacetate on its pharmacokinetics and oral bioavailability in human volunteers: A stable isotope study. Toxicol. Sci. 2006, 92, 42–50.
- Chu, X.; Schwartz, R.; Diamond, M.P.; Raju, R.P. A Combination Treatment Strategy for Hemorrhagic Shock in a Rat Model Modulates Autophagy. Front. Med. 2019, 6, 281.
- Kumar, A.; Kant, S.; Singh, S.M. Novel molecular mechanisms of antitumor action of dichloroacetate against T cell lymphoma: Implication of altered glucose metabolism, pH homeostasis and cell survival regulation. Chem. Biol. Interact. 2012, 199, 29–37.
- Norberg, E.; Lako, A.; Chen, P.H.; Stanley, I.A.; Zhou, F.; Ficarro, S.B.; Chapuy, B.; Chen, L.; Rodig, S.; Shin, D.; et al. Differential contribution of the mitochondrial translation pathway to the survival of diffuse large B-cell lymphoma subsets. Cell Death Differ. 2017, 24, 251–262.
- Grassian, A.R.; Parker, S.J.; Davidson, S.M.; Divakaruni, A.S.; Green, C.R.; Zhang, X.; Slocum, K.L.; Pu, M.; Lin, F.; Vickers, C.; et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014, 74, 3317–3331.
- Turcan, S.; Fabius, A.W.; Borodovsky, A.; Pedraza, A.; Brennan, C.; Huse, J.; Viale, A.; Riggins, G.J.; Chan, T.A. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget 2013, 4, 1729–1736.
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784.
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012, 15, 110–121.
- Sugimoto, K.; Suzuki, H.I.; Fujimura, T.; Ono, A.; Kaga, N.; Isobe, Y.; Sasaki, M.; Taka, H.; Miyazono, K.; Komatsu, N. A clinically attainable dose of L-asparaginase targets glutamine addiction in lymphoid cell lines. Cancer Sci. 2015, 106, 1534–1543.
- Nam, S.J.; Kim, S.; Paik, J.H.; Kim, T.M.; Heo, D.S.; Kim, C.W.; Jeon, Y.K. An increase in indoleamine 2,3-dioxygenase-positive cells in the tumor microenvironment predicts favorable prognosis in patients with diffuse large B-cell lymphoma treated with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone. Leuk. Lymphoma 2016, 57, 1956–1960.
- Gelebart, P.; Zak, Z.; Anand, M.; Belch, A.; Lai, R. Blockade of fatty acid synthase triggers significant apoptosis in mantle cell lymphoma. PLoS ONE 2012, 7, e33738.
- Dengler, M.A.; Weilbacher, A.; Gutekunst, M.; Staiger, A.M.; Vöhringer, M.C.; Horn, H.; Ott, G.; Aulitzky, W.E.; van der Kuip, H. Discrepant NOXA (PMAIP1) transcript and NOXA protein levels: A potential Achilles’ heel in mantle cell lymphoma. Cell Death Dis. 2014, 5, e1013.
- Bastos, D.C.; Paupert, J.; Maillard, C.; Seguin, F.; Carvalho, M.A.; Agostini, M.; Coletta, R.D.; Noël, A.; Graner, E. Effects of fatty acid synthase inhibitors on lymphatic vessels: An in vitro and in vivo study in a melanoma model. Lab. Investig. 2017, 97, 194–206.
- Uddin, S.; Hussain, A.R.; Ahmed, M.; Bu, R.; Ahmed, S.O.; Ajarim, D.; Al-Dayel, F.; Bavi, P.; Al-Kuraya, K.S. Inhibition of fatty acid synthase suppresses c-Met receptor kinase and induces apoptosis in diffuse large B-cell lymphoma. Mol. Cancer Ther. 2010, 9, 1244–1255.
- Bhatt, A.P.; Jacobs, S.R.; Freemerman, A.J.; Makowski, L.; Rathmell, J.C.; Dittmer, D.P.; Damania, B. Dysregulation of fatty acid synthesis and glycolysis in non-Hodgkin lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, 11818–11823.
- Molyneux, E.; Merrick, B.; Khanim, F.L.; Banda, K.; Dunn, J.A.; Iqbal, G.; Bunce, C.M.; Drayson, M.T. Bezafibrate and medroxyprogesterone acetate in resistant and relapsed endemic Burkitt lymphoma in Malawi; an open-label, single-arm, phase 2 study (ISRCTN34303497). Br. J. Haematol. 2014, 164, 888–890.
- Huang, J.; Das, S.K.; Jha, P.; Al Zoughbi, W.; Schauer, S.; Claudel, T.; Sexl, V.; Vesely, P.; Birner-Gruenberger, R.; Kratky, D.; et al. The PPARα agonist fenofibrate suppresses B-cell lymphoma in mice by modulating lipid metabolism. BioChim. Biophys. Acta 2013, 1831, 1555–1565.
- Xiong, J.; Bian, J.; Wang, L.; Zhou, J.Y.; Wang, Y.; Zhao, Y.; Wu, L.L.; Hu, J.J.; Li, B.; Chen, S.J.; et al. Dysregulated choline metabolism in T-cell lymphoma: Role of choline kinase-α and therapeutic targeting. Blood Cancer J. 2015, 5, 287.
- Shrestha, P.; Davis, D.A.; Veeranna, R.P.; Carey, R.F.; Viollet, C.; Yarchoan, R. Hypoxia-inducible factor-1 alpha as a therapeutic target for primary effusion lymphoma. PLoS Pathog. 2017, 13, e1006628.
- Bhalla, S.; Evens, A.M.; Prachand, S.; Schumacker, P.T.; Gordon, L.I. Paradoxical regulation of hypoxia inducible factor-1α (HIF-1α) by histone deacetylase inhibitor in diffuse large B-cell lymphoma. PLoS ONE 2013, 8, e81333.
- Yang, B.; Yu, D.; Liu, J.; Yang, K.; Wu, G.; Liu, H. Antitumor activity of SAHA, a novel histone deacetylase inhibitor, against murine B cell lymphoma A20 cells in vitro and in vivo. Tumour Biol. 2015, 36, 5051–5061.
- Jung, K.Y.; Wang, H.; Teriete, P.; Yap, J.L.; Chen, L.; Lanning, M.E.; Hu, A.; Lambert, L.J.; Holien, T.; Sundan, A.; et al. Perturbation of the c-Myc-Max protein-protein interaction via synthetic α-helix mimetics. J. Med. Chem. 2015, 58, 3002–3024.
- Huang, M.J.; Cheng, Y.C.; Liu, C.R.; Lin, S.; Liu, H.E. A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp. Hematol. 2006, 34, 1480–1489.
- Whitfield, J.R.; Beaulieu, M.E.; Soucek, L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10.
- Broecker-Preuss, M.; Becher-Boveleth, N.; Bockisch, A.; Dührsen, U.; Müller, S. Regulation of glucose uptake in lymphoma cell lines by c-MYC- and PI3K-dependent signaling pathways and impact of glycolytic pathways on cell viability. J. Transl. Med. 2017, 15, 158.
- Erdmann, T.; Klener, P.; Lynch, J.T.; Grau, M.; Vočková, P.; Molinsky, J.; Tuskova, D.; Hudson, K.; Polanska, U.M.; Grondine, M.; et al. Sensitivity to PI3K and AKT inhibitors is mediated by divergent molecular mechanisms in subtypes of DLBCL. Blood 2017, 130, 310–322.
- Mediani, L.; Gibellini, F.; Bertacchini, J.; Frasson, C.; Bosco, R.; Accordi, B.; Basso, G.; Bonora, M.; Calabrò, M.L.; Mattiolo, A.; et al. Reversal of the glycolytic phenotype of primary effusion lymphoma cells by combined targeting of cellular metabolism and PI3K/Akt/ mTOR signaling. Oncotarget 2016, 7, 5521–5537.
- Yahiaoui, A.; Meadows, S.A.; Sorensen, R.A.; Cui, Z.H.; Keegan, K.S.; Brockett, R.; Chen, G.; Quéva, C.; Li, L.; Tannheimer, S.L. PI3Kδ inhibitor idelalisib in combination with BTK inhibitor ONO/GS-4059 in diffuse large B cell lymphoma with acquired resistance to PI3Kδ and BTK inhibitors. PLoS ONE 2017, 12, e0171221.
- Ferreira, A.C.; Robaina, M.C.; Rezende, L.M.; Severino, P.; Klumb, C.E. Histone deacetylase inhibitor prevents cell growth in Burkitt’s lymphoma by regulating PI3K/Akt pathways and leads to upregulation of miR-143, miR-145, and miR-101. Ann. Hematol. 2014, 93, 983–993.
- Argyriou, P.; Papageorgiou, S.G.; Panteleon, V.; Psyrri, A.; Bakou, V.; Pappa, V.; Spathis, A.; Economopoulou, P.; Papageorgiou, E.; Economopoulos, T.; et al. Hypoxia-inducible factors in mantle cell lymphoma: Implication for an activated mTORC1→HIF-1α pathway. Ann. Hematol. 2011, 90, 315–322.
- Kittipongdaja, W.; Wu, X.; Garner, J.; Liu, X.; Komas, S.M.; Hwang, S.T.; Schieke, S.M. Rapamycin Suppresses Tumor Growth and Alters the Metabolic Phenotype in T-Cell Lymphoma. J. Investig. Dermatol 2015, 135, 2301–2308.
- Sekihara, K.; Saitoh, K.; Han, L.; Ciurea, S.; Yamamoto, S.; Kikkawa, M.; Kazuno, S.; Taka, H.; Kaga, N.; Arai, H.; et al. Targeting mantle cell lymphoma metabolism and survival through simultaneous blockade of mTOR and nuclear transporter exportin-1. Oncotarget 2017, 8, 34552–34564.
- Bhatt, A.P.; Bhende, P.M.; Sin, S.H.; Roy, D.; Dittmer, D.P.; Damania, B. Dual inhibition of PI3K and mTOR inhibits autocrine and paracrine proliferative loops in PI3K/Akt/mTOR-addicted lymphomas. Blood 2010, 115, 4455–4463.
- Tarantelli, C.; Gaudio, E.; Arribas, A.J.; Kwee, I.; Hillmann, P.; Rinaldi, A.; Cascione, L.; Spriano, F.; Bernasconi, E.; Guidetti, F.; et al. PQR309 Is a Novel Dual PI3K/mTOR Inhibitor with Preclinical Antitumor Activity in Lymphomas as a Single Agent and in Combination Therapy. Clin. Cancer Res. 2018, 24, 120–129.
- Montraveta, A.; Xargay-Torrent, S.; López-Guerra, M.; Rosich, L.; Pérez-Galán, P.; Salaverria, I.; Beà, S.; Kalko, S.G.; de Frias, M.; Campàs, C.; et al. Synergistic anti-tumor activity of acadesine (AICAR) in combination with the anti-CD20 monoclonal antibody rituximab in in vivo and in vitro models of mantle cell lymphoma. Oncotarget 2014, 5, 726–739.
- Choudhary, G.S.; Al-Harbi, S.; Mazumder, S.; Hill, B.T.; Smith, M.R.; Bodo, J.; Hsi, E.D.; Almasan, A. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015, 6, e1593.
- Campàs, C.; Santidrián, A.F.; Domingo, A.; Gil, J. Acadesine induces apoptosis in B cells from mantle cell lymphoma and splenic marginal zone lymphoma. Leukemia 2005, 19, 292–294.
- Campàs, C.; Lopez, J.M.; Santidrián, A.F.; Barragán, M.; Bellosillo, B.; Colomer, D.; Gil, J. Acadesine activates AMPK and induces apoptosis in B-cell chronic lymphocytic leukemia cells but not in T lymphocytes. Blood 2003, 101, 3674–3680.
- Santidrián, A.F.; González-Gironès, D.M.; Iglesias-Serret, D.; Coll-Mulet, L.; Cosialls, A.M.; de Frias, M.; Campàs, C.; González-Barca, E.; Alonso, E.; Labi, V.; et al. AICAR induces apoptosis independently of AMPK and p53 through up-regulation of the BH3-only proteins BIM and NOXA in chronic lymphocytic leukemia cells. Blood 2010, 116, 3023–3032.
- Sengupta, T.K.; Leclerc, G.M.; Hsieh-Kinser, T.T.; Leclerc, G.J.; Singh, I.; Barredo, J.C. Cytotoxic effect of 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) on childhood acute lymphoblastic leukemia (ALL) cells: Implication for targeted therapy. Mol. Cancer 2007, 6, 46.
- Shi, W.Y.; Xiao, D.; Wang, L.; Dong, L.H.; Yan, Z.X.; Shen, Z.X.; Chen, S.J.; Chen, Y.; Zhao, W.L. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis. 2012, 3, e275.
- Rosilio, C.; Lounnas, N.; Nebout, M.; Imbert, V.; Hagenbeek, T.; Spits, H.; Asnafi, V.; Pontier-Bres, R.; Reverso, J.; Michiels, J.F.; et al. The metabolic perturbators metformin, phenformin and AICAR interfere with the growth and survival of murine PTEN-deficient T cell lymphomas and human T-ALL/T-LL cancer cells. Cancer Lett. 2013, 336, 114–126.
- Offman, M.N.; Krol, M.; Patel, N.; Krishnan, S.; Liu, J.; Saha, V.; Bates, P.A. Rational engineering of L-asparaginase reveals importance of dual activity for cancer cell toxicity. Blood 2011, 117, 1614–1621.
- Abd-Aziz, N.; Stanbridge, E.J.; Shafee, N. Bortezomib attenuates HIF-1- but not HIF-2-mediated transcriptional activation. Oncol. Lett. 2015, 10, 2192–2196.
- Bhatt, R.; Ravi, D.; Evens, A.M.; Parekkadan, B. Scaffold-mediated switching of lymphoma metabolism in culture. Cancer Metab. 2022, 10, 15.
- Welcker, M.; Orian, A.; Jin, J.; Grim, J.E.; Harper, J.W.; Eisenman, R.N.; Clurman, B.E. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. USA 2004, 101, 9085–9090.
- Bahram, F.; von der Lehr, N.; Cetinkaya, C.; Larsson, L.G. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood 2000, 95, 2104–2110.
- Popov, N.; Wanzel, M.; Madiredjo, M.; Zhang, D.; Beijersbergen, R.; Bernards, R.; Moll, R.; Elledge, S.J.; Eilers, M. The ubiquitin-specific protease USP28 is required for MYC stability. Nat. Cell Biol. 2007, 9, 765–774.
- Xie, H.; Tang, C.H.; Song, J.H.; Mancuso, A.; Del Valle, J.R.; Cao, J.; Xiang, Y.; Dang, C.V.; Lan, R.; Sanchez, D.J.; et al. IRE1α RNase-dependent lipid homeostasis promotes survival in Myc-transformed cancers. J. Clin. Investig. 2018, 128, 1300–1316.
- Calvo-Vidal, M.N.; Zamponi, N.; Krumsiek, J.; Stockslager, M.A.; Revuelta, M.V.; Phillip, J.M.; Marullo, R.; Tikhonova, E.; Kotlov, N.; Patel, J.; et al. Oncogenic HSP90 Facilitates Metabolic Alterations in Aggressive B-cell Lymphomas. Cancer Res. 2021, 81, 5202–5216.
- Dreyling, M.; Santoro, A.; Mollica, L.; Leppä, S.; Follows, G.A.; Lenz, G.; Kim, W.S.; Nagler, A.; Panayiotidis, P.; Demeter, J.; et al. Phosphatidylinositol 3-Kinase Inhibition by Copanlisib in Relapsed or Refractory Indolent Lymphoma. J. Clin. Oncol. 2017, 35, 3898–3905.
- Horwitz, S.M.; Koch, R.; Porcu, P.; Oki, Y.; Moskowitz, A.; Perez, M.; Myskowski, P.; Officer, A.; Jaffe, J.D.; Morrow, S.N.; et al. Activity of the PI3K-δ,γ inhibitor duvelisib in a phase 1 trial and preclinical models of T-cell lymphoma. Blood 2018, 131, 888–898.
- Gu, L.; Gao, J.; Li, Q.; Zhu, Y.P.; Jia, C.S.; Fu, R.Y.; Chen, Y.; Liao, Q.K.; Ma, Z. Rapamycin reverses NPM-ALK-induced glucocorticoid resistance in lymphoid tumor cells by inhibiting mTOR signaling pathway, enhancing G1 cell cycle arrest and apoptosis. Leukemia 2008, 22, 2091–2096.
- Gu, L.; Xie, L.; Zuo, C.; Ma, Z.; Zhang, Y.; Zhu, Y.; Gao, J. Targeting mTOR/p70S6K/glycolysis signaling pathway restores glucocorticoid sensitivity to 4E-BP1 null Burkitt Lymphoma. BMC Cancer 2015, 15, 529.
- Drew, B.G.; Kingwell, B.A. Acadesine, an adenosine-regulating agent with the potential for widespread indications. Expert Opin. Pharmacother. 2008, 9, 2137–2144.
- Van Den Neste, E.; Van den Berghe, G.; Bontemps, F. AICA-riboside (acadesine), an activator of AMP-activated protein kinase with potential for application in hematologic malignancies. Expert Opin. Investig. Drugs 2010, 19, 571–578.
- Vakana, E.; Platanias, L.C. AMPK in BCR-ABL expressing leukemias. Regulatory effects and therapeutic implications. Oncotarget 2011, 2, 1322–1328.
- Liu, X.; Chhipa, R.R.; Pooya, S.; Wortman, M.; Yachyshin, S.; Chow, L.M.; Kumar, A.; Zhou, X.; Sun, Y.; Quinn, B.; et al. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc. Natl. Acad. Sci. USA 2014, 111, E435–E444.
- Ly, P.; Kim, S.B.; Kaisani, A.A.; Marian, G.; Wright, W.E.; Shay, J.W. Aneuploid human colonic epithelial cells are sensitive to AICAR-induced growth inhibition through EGFR degradation. Oncogene 2013, 32, 3139–3146.
- Jose, C.; Bellance, N.; Chatelain, E.H.; Benard, G.; Nouette-Gaulain, K.; Rossignol, R. Antiproliferative activity of levobupivacaine and aminoimidazole carboxamide ribonucleotide on human cancer cells of variable bioenergetic profile. Mitochondrion 2012, 12, 100–109.
- Rattan, R.; Giri, S.; Singh, A.K.; Singh, I. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. J. Biol. Chem. 2005, 280, 39582–39593.
- Van Den Neste, E.; Cazin, B.; Janssens, A.; González-Barca, E.; Terol, M.J.; Levy, V.; Pérez de Oteyza, J.; Zachee, P.; Saunders, A.; de Frias, M.; et al. Acadesine for patients with relapsed/refractory chronic lymphocytic leukemia (CLL): A multicenter phase I/II study. Cancer Chemo. Ther. Pharmacol. 2013, 71, 581–591.
- Wang, H.; Shao, R.; Liu, W.; Tang, H.; Lu, Y. Identification of a prognostic metabolic gene signature in diffuse large B-cell lymphoma. J. Cell Mol. Med. 2021, 25, 7066–7077.
- He, J.; Chen, Z.; Xue, Q.; Sun, P.; Wang, Y.; Zhu, C.; Shi, W. Identification of molecular subtypes and a novel prognostic model of diffuse large B-cell lymphoma based on a metabolism-associated gene signature. J. Transl. Med. 2022, 20, 186.
- Zhang, J.; Medina-Cleghorn, D.; Bernal-Mizrachi, L.; Bracci, P.M.; Hubbard, A.; Conde, L.; Riby, J.; Nomura, D.K.; Skibola, C.F. The potential relevance of the endocannabinoid, 2-arachidonoylglycerol, in diffuse large B-cell lymphoma. Oncoscience 2016, 3, 31–41.
- Yang, F.; Du, J.; Zhang, H.; Ruan, G.; Xiang, J.; Wang, L.; Sun, H.; Guan, A.; Shen, G.; Liu, Y.; et al. Serum Metabolomics of Burkitt Lymphoma Mouse Models. PLoS ONE 2017, 12, e0170896.
- Stenson, M.; Pedersen, A.; Hasselblom, S.; Nilsson-Ehle, H.; Karlsson, B.G.; Pinto, R.; Andersson, P.O. Serum nuclear magnetic resonance-based metabolomics and outcome in diffuse large B-cell lymphoma patients-a pilot study. Leuk. Lymphoma 2016, 57, 1814–1822.
- Le, Y.; Shen, X.; Kang, H.; Wang, Q.; Li, K.; Zheng, J.; Yu, Y. Accelerated, untargeted metabolomics analysis of cutaneous T-cell lymphoma reveals metabolic shifts in plasma and tumor adjacent skins of xenograft mice. J. Mass Spectrom 2018, 53, 739.
- Eberlin, L.S.; Gabay, M.; Fan, A.C.; Gouw, A.M.; Tibshirani, R.J.; Felsher, D.W.; Zare, R.N. Alteration of the lipid profile in lymphomas induced by MYC overexpression. Proc. Natl. Acad. Sci. USA 2014, 111, 10450–10455.


