1. Therapeutic Strategies Targeting Metabolism
In the past decade, progress in targeting lymphoma metabolism therapeutically has been limited. Only a few metabolism-based drugs for lymphoma have been successfully developed, including those which already have been approved for clinical application and ongoing clinical trials (Table 1).
Table 1. Primary approved drugs and ongoing clinical trials targeting lymphoma metabolism.
| Agent |
Target |
Mechanism |
Type of Diseases |
| methotrexate |
DHFR |
folate to THF conversion |
prophylaxis and treatment of CNS lymphoma |
| IM156 |
mitochondrial complex I inhibitor |
mitochondrial oxidative phosphorylation and NADH oxidation |
lymphomas (NCT03272256) |
| IACS-010759 |
oxidative phosphorylation inhibitor |
mitochondrial oxidative phosphorylation and NADH oxidation |
R/R AML (NCT02882321) |
| AZD-3965 |
MCT1 |
mediate the bidirectional transport of lactatein and out of cells |
DLBCL, BL (NCT01791595) |
| CPI-613 |
mitochondria |
oxidative metabolism |
T-cell NHL (NCT04217317), R/R BL or HGBCL (NCT03793140) |
| IM156 |
mitochondria |
complex I |
advanced lymphoma (NCT03272256) |
| IACS-010759 |
mitochondria |
complex I |
R/R AML (NCT02882321) |
| ibrutinib |
BTK inhibitor |
downstream pro-proliferative kinase of BCR signal |
MCL, CLL/SLL, WM, MZL, cGVHD (NCT02169180, NCT04771507, NCT02604511, NCT04212013, NCT05348096) |
| acalabrutinib |
MCL, CLL/SLL, MZL (NCT02213926, NCT04505254, NCT04646395) |
| rapalogs |
mTORC1 inhibitor |
rapamycin analogues |
MCL |
| temsirolimus |
mTORC1 inhibitor |
cell cycle arrest in the G1 phase, inhibits tumor angiogenesis by reducing synthesis of VEGF |
MCL (NCT01078142, NCT01180049), HL (NCT01902160), R/R NHL (NCT01281917) |
| idelalisib |
PI3Kδ inhibitor |
against PI3Kδ isoforms |
iNHL (NCT01282424), HL (NCT01393106), FL (NCT03568929), CLL (NCT03582098) |
| copanlisib |
PI3Kδ and PI3Kα inhibitor |
against PI3K-α and PI3K-δ isoforms |
iNHL, DLBCL, MCL, PTCL (NCT05217914, NCT04433182, NCT04939272, NCT03877055, NCT03052933) |
| duvelisib |
PI3Kδ and PI3Kγinhibitor |
against PI3Kδ and PI3Kγ isoforms |
CLL/SLL, DLBCL, PTCL (NCT02004522, NCT04890236, NCT04803201) |
| umbralisib |
PI3Kδ and casein kinase-1 epsilon inhibitor |
against PI3Kδisoforms and casein kinase-1 epsilon |
MZL, FL, CLL, WM (NCT03919175, NCT03364231, NCT02535286) |
| L-asparaginase |
asparagine |
inhibit protein biosynthesis in lymphoblasts |
ALL (NCT01518517, NCT00506597), NK/T cell lymphoma (NCT00854425) |
| Telaglenastat (CB-839) |
glutaminase inhibitor |
glutamine to glutamate conversion |
NHL (NCT02071888), including MCL, WM, TCL |
| AG-270 |
MAT2A |
production of S-adenosylmethionine |
advanced lymphoma (NCT03361358, NCT03435250) |
| Devimistat (CPI-613) |
lipoate analog |
mitochondrial oxidative metabolism |
R/R T-cell NHL (NCT04217317) |
1.1. Targeting Metabolic Pathways
1.1.1. Targeting Glucose Metabolism
Pharmaceutical inhibition of glycolytic enzymes may provide a novel therapeutic strategy for NHLs. HK2 induces the conversion of glucose to glucose-6-phosphate, which is the first step in glycolysis. Two-deoxyglucose (2-DG), an analog of glucose, once bound and phosphorylated by HK2, is not further metabolized, thereby suppressing HK2 [
191]. In NHL cell lines, 2-DG can inhibit cell proliferation both under hypoxia and normoxia (
Table 2). When combined with methylprednisolone, 2-DG synergistically inhibits cell proliferation by downregulating HIF-1α and c-Myc [
192]. Because 2-DG-induced toxicity is regulated by members of the BCL2 family, it can be enhanced by antagonizing BCL2-positive lymphoma cell lines. 2-DG can also induce GADD153/CHOP expression, which is a marker of endoplasmic reticulum stress and an activator of Bim [
193]. A glycolysis inhibitor that targets Mcl-1 can restore the sensitivity of lymphoma cells (primary Ramos cells or Eµ-myc) to ABT-737-induced apoptosis [
52]. In MCL cells, 2-DG and glucose restriction via anti-glycolytic drugs inhibit TRAIL-induced cell death, indicating that mitochondrial metabolism directly regulates the sensitivity of tumor cells to apoptosis [
194]. 3-bromopyruvate (3-BrPA) cannot only restrain tumor glycolysis acting through the hexokinase step, but also hampers mitochondrial ATP production. Both in vitro and in vivo, 3-BrPA demonstrated a significant positive tumor response in Raji-lymphoma-bearing mice [
195,
196] (
Table 2). With augmented induction of apoptosis, 3-bromopyruvate (3-BP), a pyruvate inhibitor and brominated derivative of pyruvate, can inhibit metabolism and survival in Dalton’s lymphoma (DL) cells [
197] (
Table 2). FX11, a competitive small-molecule inhibitor of LDHA, inhibits cell proliferation and induces death in P493 BL cells by reducing ATP levels and inducing significant oxidative stress [
198,
199] (
Table 2). Interestingly, in lymphoma-bearing mice, α-tocopherol, the most active component of vitamin E, also contributes to keeping cell proliferation in check by downregulating LDHA, PKC-α, and c-Myc expression [
199] (
Table 2). Inhibition of MCT1 using AZD3965 that blocked lactate efflux led to the accumulation of glycolytic intermediates in vitro and significant downregulation of tumor proliferation in vivo in the Raji BL model. Moreover, when combined with doxorubicin or rituximab, enhanced cell proliferation inhibition and cell death were observed in vitro and in vivo [
200,
201]. Dichochloroacetate (DCA), a by-product of drinking water disinfection, blocks phosphorylation of PDK at the mitochondrial membrane level, and thus, glycolysis is downregulated due to the activation of the PDH [
202,
203] (
Table 2). Accompanied by modulation of glycolysis and expression of HIF1-α, DCA inhibited cell survival in a DL mouse model [
204]. Taken together, these data suggest that glycolytic enzymes with deregulated expression levels are potential biomarkers and have become potential therapeutic targets in NHLs. Tigecycline, approved by FDA, is selectively toxic to OxPhos-DLBCL cell lines and primary tumors and can pharmacologically disturb the mitochondrial translation pathway (
Table 2). These findings indicate that the mitochondrial translation pathway is a potential therapeutic target for these tumors as well [
56].
The presence of IDH1 or IDH2 mutations and accumulation of 2-HG may result in dependence on specific pathways and the introduction of therapeutic vulnerability. For instance, tumors that harbor IDH mutations are more sensitive to electron-transport-chain inhibitors [
205], hypomethylating agents [
190], depletion of the canonical coenzyme NAD
+, and chemoradiotherapy [
206]. Pharmacological molecules targeting mutant IDH1 and IDH2 enzymes are being explored and assessed for antitumor efficacy.
Table 2. Targeting lymphoma metabolism via metabolic enzymes, metabolite depletion, and/or signaling pathways.
| Pathway |
Compound |
Application |
Development Stage |
Ref |
| HK2 |
2-DG |
B-NHL |
cell lines |
[192] |
| 3-BrPA |
BL |
cell lines, mice |
[195,196] |
| Pyruvate |
3-BP |
T-NHL |
mice (DL) |
[197] |
| LDHA |
FX11 |
BL |
cell line |
[198,199] |
| α-Tocopherol |
T-NHL |
mice (DL) |
[199] |
| PDK |
DCA |
DL |
mice |
[202,203] |
| Mitochondrial protein translation |
Tigecycline |
OxPhos-DLBCL cell lines |
FDA-approved |
[56] |
| Glutaminase |
BPTES |
BL |
mice |
[47] |
| Glutamine uptake |
L-asparaginase |
NHL |
cell lines |
[207] |
| SHMT1/2 |
SHIN1 |
DLBCL |
cell lines |
[33] |
| FASN |
orlistat |
MCL |
cell lines |
[86,208] |
| T-NHL |
cell lines, mice |
[209] |
| C75 |
DLBCL, PEL, and B-NHL |
cell lines |
[84,85] |
| NA |
BaP |
BL |
patients |
[210] |
| PPARα |
Fenofibrate |
B-NHL |
mice |
[211] |
| Choline kinase |
CK37 |
T-NHL |
mice |
[42] |
| HIF-1α |
PX-478 |
PEL |
cell lines |
[115] |
| PCI-24781(HDACi) |
DLBCL |
Phase 1/2 |
[212] |
| SAHA(HDACi) |
B-NHL |
cell lines, mice |
[213] |
| MYC |
10058-F4 |
DLBCL |
cell lines |
[214,215,216] |
| PI3K |
LY294002 |
B-NHL |
cell lines |
[217] |
| |
AZD8835 |
ABC-DLBCL |
cell lines |
[218] |
| AKT |
Akti1/2 |
PEL |
cell lines |
[219] |
| AZD-5363 |
PTEN-deficient DLBCL |
cell lines, mice |
[218] |
| MK-2206 |
ABC-DLBCL |
mice |
[220] |
| NaB (HDACi) |
BL |
cell lines |
[221] |
| mTOR |
Rapamycin |
ALCL, NHL |
cell lines |
[36,131] |
| mTOR C1/2 |
AZD-2014 |
MCL |
cell lines |
[222] |
| Dual inhibitor of PI3K and mTOR |
NVP-BEZ235 |
PEL |
mice |
[223] |
| PF-04091502 |
PEL |
cell lines |
[219] |
| Bimiralisib (PQR309) |
DLBCL, MCL, SMZL, CLL, HL, and ALCL |
cell lines, mice |
[224] |
| AMPK |
AICAR |
MCL, SMZL, FL, and CLL |
cell lines, mice and patients |
[159,225,226,227,228,229] |
| Metformin |
B and T-NHL |
cell lines, mice |
[157,230] |
| Phenformin |
PTEN-deficient T-cell lymphomas |
cell lines |
[230] |
1.1.2. Targeting Amino Acid Metabolism
Glutamine metabolism plays a crucial role in cell survival and proliferation under glucose deficiency and hypoxia, making it sensitive to the glutaminase inhibitor Bis-2-[5-(phenylacetamido)-1,3,4-thiadiazol-2-yl]ethyl sulfide (BPTES) [
47] (
Table 2). L-Asparaginase (L-ASNase), which exhibits some glutaminase activity, can hydrolyze extracellular glutamine to glutamate and ammonia and prevent glutamine from entering the cell [
231]. In NHL cell lines, L-ASNase exerts cytotoxicity by depriving the cells of glutamine, resulting in the suppression of cell growth and survival [
207] (
Table 1 and
Table 2).
1.1.3. Targeting Lipid Metabolism
Pharmacological inhibition using the FASN-specific inhibitor C75 triggered caspase-dependent apoptosis in DLBCL cell lines [
84] (
Table 2). FASN, up-regulated in PEL and other types of non-viral B-NHLs in a PI3K dependent manner, is sensitive to C75 as well [
85] (
Table 2). FASN is also highly and consistently expressed in MCLs. MCL cell lines, in which FASN is highly and consistently expressed, exhibited significant apoptosis when treated with orlistat, an anti-obesity drug, which is also an inhibitor of FASN and is approved by the FDA [
86] (
Table 2). Orlistat can also interfere with the ubiquitination of NOXA protein and induce apoptosis, thereby offering new strategies to kill bortezomib-resistant MCL cells [
208]. TCLs, when treated with orlistat in vitro, manifest tumor-specific inhibition of cell survival and induction of apoptosis [
209]. Orlistat-induced tumor growth retardation decreased tumor cell survival and chemosensitization to cisplatin, and a prolonged life span was observed in tumor-bearing mice [
209] (
Table 2). BaP, a redeployed drug that combines bezafibrate and medroxyprogesterone acetate, whose complete mechanism was not fully understood, was extended to the clinic and demonstrated efficacy with low toxicity in clinical trials involving patients with BL (ISRCTN34303497) [
210] (
Table 2). Interestingly, recent findings indicate that obesity is associated with an increased risk of developing malignant lymphomas. In wild-type B-cell lymphoma mice, tumor size was significantly associated with the depletion of white adipose tissues (WAT) and elevated levels of lipid metabolites. Thus, tumor growth can be significantly suppressed with PPARα agonists and the lipid-lowering drug fenofibrate [
211] (
Table 2).
1.2. Targeting Oncogenic Regulators
1.2.1. HIF-1α Inhibitors
HIF-1α plays an important role in the pathogenesis of lymphoma. Inhibition of HIF-1α may have a therapeutic effect on lymphoma. PX-478 is a small-molecule inhibitor of HIF-1α and inhibits lymphoma cell growth even under normoxia [
115] (
Table 2). PCI-24781 is a broad-spectrum histone deacetylase inhibitor (HDACi) that can promote the accumulation of HIF-1α and induce initial autophagy of lymphoma cells. Long-term incubation with HDACi can inhibit the expression of HIF-1α [
212] (
Table 2). Suberoylanilide hydroxamic acid (SAHA, vorinostat), a second-generation HDACi, can inhibit cell proliferation and induce apoptosis by inhibiting the expression of HIF-1α in B-cell lymphoma in vitro and in vivo [
213]. Bortezomib is the first proteasome inhibitor used for treating MCLs by suppressing the transcription and expression of HIF-1-specific target genes. Although HIF-1α is not destroyed by bortezomib, heterodimeric HIF-1 cannot transactivate the target genes [
232] (
Figure 1).
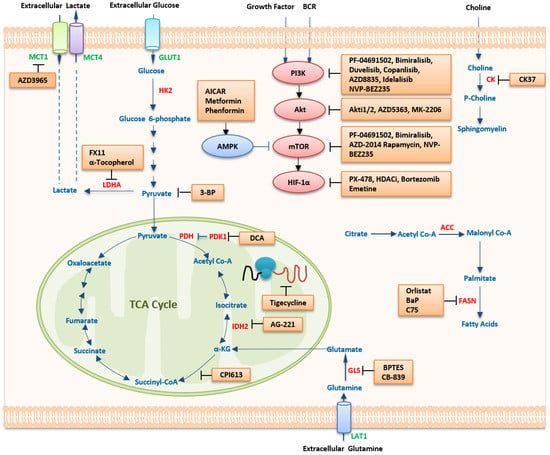
Figure 1. Therapeutic strategies targeting metabolism in lymphoma. Several pathways of the bioenergetic and anabolic metabolism of malignant cells harbor targets for the treatment of cancer. In general, agents that disrupt these pathways would be expected to result in deficiencies in energy and materials needed for cell proliferation and survival, forming the basis for their use as anticancer therapies. Abbreviations: MCT1/4, monocarboxylate transporter 1/4; GULT1, glucose transporter 1; HK2, hexokinase 2; LDHA, lactate dehydrogenase A; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; DCA, dichochloroacetate; IDH2, isocitrate dehydrogenase; α-KG, α-ketoglutarate; TCA, tricarboxylic acid; GLS, glutaminase; BPTES, bis-2-(5-phenylacetamido-1,2,4-diathiazol-2-yl) ethyl sulfide; LAT1, L-type amino-acid transporter 1; FASN, fatty acid synthase; ACC, acetyl-CoA carboxylase; HDACi, histone deacetylase inhibitor; HIF-1α, hypoxia-inducible factor 1 alpha; mTOR, mammalian target of rapamycin; AMPK, 5ʹ-AMP-activated protein kinase; AICAR, 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside; PI3K, phosphatidylinositol 3-kinase; BCR, B cell receptor; CK, choline kinase. In the metabolic pathways, transporters are in green color, key metabolic enzymes are in red color, metabolites are in blue color, and inhibitors are in black color.
1.2.2. MYC Inhibitors
Most MYC inhibitors are intended to interfere with the MYC–MAX interaction. Because MYC lacks hydrophobic pockets, its structure makes it difficult to target the MYC protein directly. Therefore, inhibition of c-Myc is an attractive strategy to inhibit glucose metabolism and the proliferation of lymphoma cells. In DLBCL cell lines, including GCB and ABC, cell proliferation was inhibited by the c-Myc inhibitor 10058-F4, as a single agent and in combination with 2-DG [
217]. However, 10058-F4 is ineffective in vivo due to its poor bioavailability and rapid metabolism and clearance in other cancers [
214,
215,
216] (
Table 2). Thus, no small-molecule inhibitor targeting the
MYC gene is currently tested in clinical trials. Recently, Bhatt et al. cultured DLBCL cells in three-dimensional matrices and discovered a reduced proliferative activity and altered metabolism that is consistent with in vivo tumor cell function [
233]. Development of three-dimensional conditions that stabilize tumor cell function towards in vivo phenotype might be helpful for the understanding of lymphoma metabolic process and make progress in relevant research.
A therapeutic approach targeting MYC phosphorylation and degradation is a promising way of treating cancers addicted to high MYC protein levels [
234,
235,
236]. The enzyme 1α/X-box binding protein 1/stearoyl-CoA-desaturase 1 (IRE1α/XBP1/SCD1) axis plays a protective role in counterbalancing anabolism mediated by MYC overexpression. Genetic and pharmacological inhibition of XBP1 leads to MYC-dependent apoptosis, which can be alleviated by exogenous unsaturated fatty acids. In addition, IRE1α inhibition can enhance the cytotoxic effects of standard chemotherapy against BL with MYC overexpression [
237].
The oncogenic form of HSP90 optimizes several MYC metabolic pathways, including the production of nucleotides [
121]. Given the importance of MYC in driving metabolic reprogramming in lymphoma progress, oncogenic HSP90 inhibitors could reverse the immunosuppressive effects on the lymphoma microenvironment and thus potentially improve efficacy of lymphoma immunotherapy.
1.2.3. Targeting The PI3K/mTOR Pathway
PI3K Inhibitors
Treatment with the PI3K inhibitor LY294002, either as a single agent or in combination with 2-DG, reduces cell survival, FDG uptake, and cell growth [
217] (
Table 2). Inhibition of PI3K-α and -δ by copanlisib demonstrated manageable safety and significant efficacy in relapsed or refractory (R/R) indolent lymphoma patients who were heavily pretreated. High response rates were associated with increased expression of PI3K/BCR signaling pathway genes [
238] (
Table 1). Another PI3K-α/δ inhibitor, AZD8835, demonstrated remarkable potency in ABC-like DLBCL models by inhibiting NF-κB signaling and had a synergistic effect with the Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib both in vitro and in vivo [
218] (
Table 2). A similar anti-tumor effect was observed in the ABC DLBCL mouse xenograft model treated with a combination of the PI3Kδ inhibitor idelalisib and BTK inhibitor ONO/GS-4059 [
220]. Duvelisib, an oral inhibitor of PI3K-δ/γ isoforms, demonstrated acceptable safety and promising clinical activity in patients with R/R TCLs [
239]. In vitro, phospho-AKT (pAKT) showed a synergistic effect with duvelisib in TCL cell lines [
239] (
Table 1 and
Table 2,
Figure 1).
Akt Inhibitors
Akti 1/2, an AKT inhibitor that reduces the rate of lactate production in hypoxic conditions, displays improved cytotoxicity to PEL cells [
219]. In combination with the 2-DG, Akti 1/2 demonstrated strong synergistic cytotoxicity toward PEL cells and shifted metabolism from aerobic glycolysis toward oxidative respiration [
219] (
Table 2). Through the downregulation of MYC, the AKT inhibitor AZD5363 induced apoptosis in PTEN-deficient DLBCLs [
218] (
Table 2). Combined with the PI3Kδ inhibitor idelalisib, the AKT inhibitor MK-2206 could increase the sensitivity of tumor cells to idelalisib in an ABC DLBCL mouse xenograft [
220] (
Table 2). In BL cell lines, by regulating AKT phosphorylation and MYC protein expression, cell proliferation was inhibited by HDACi (sodium butyrate, NaB) combined with VP-16, which indicates that the PI3K/AKT pathway is a target of HDACi as well [
221] (
Table 2). Hypoglycemic agents, such as metformin, phenformin, and the AMPK activator acadesine, have strong antitumor effects on T-cell specific PTEN-deficient (tPTEN−/−) lymphoma cells [
230] (
Figure 1).
mTOR Inhibitors
Rapamycin inhibits the growth of TCLs by decreasing the glycolytic rate and glucose utilization both in vitro and in vivo. Rapamycin-treated cells displayed reduced sensitivity to low-glucose conditions but continued to rely on OXPHOS, which can be reversed by an OXPHOS inhibitor [
131] (
Table 2). Besides, accumulating evidence indicates that rapamycin may serve as a potential glucocorticoid (GC) sensitizer in lymphomas through genetic prevention of 4E-BP1 phosphorylation [
240,
241]. Rapamycin also led to a reduction in GLUT1 mRNA expression, FDG uptake, and cell survival of NHL cell lines [
36] (
Table 2). A dual mTORC1/2 kinase inhibitor AZD-2014 can inhibit cellular energy metabolism via the TCA cycle and further inhibit glycolysis in MCL cells [
222] (
Table 2,
Figure 1).
Dual Inhibitors of PI3K and mTOR
Dual targeting of PI3K and mTOR by the inhibitor NVP-BEZ235 led to improved results compared to the application of rapamycin alone in PEL [
223] (
Table 2). PF-04691502, another dual inhibitor of PI3K and mTOR, which has a synergistic effect with 2-DG, demonstrated increased cytotoxicity in PEL cells under hypoxic conditions or in a glycolytic phenotype [
219] (
Table 1).
Bimiralisib (PQR309), an oral, novel, selective dual PI3K/mTOR inhibitor, had anti-lymphoma activity as a single agent or in combination with ibrutinib, lenalidomide, and rituximab in vitro. Increased transcripts coding for the BCR pathway can improve the activity of bimiralisib. Bimiralisib showed activity in lymphoma cells with primary or secondary resistance to idelalisib and appeared to be a novel and promising therapeutic compound [
224] (
Table 2).
1.2.4. AMPK Activators
Activation of AMPK may represent an important therapeutic strategy for lymphoma treatment due to its essential role in its pathway [
242,
243,
244]. Metformin, belonging to the biguanide class of oral hypoglycemic agents, could potentially inhibit cell growth in lymphomas as an AMPK activator both in vitro and in vivo (
Table 2). The activation of AMPK promoted by metformin was associated with inhibition of the mTOR pathway without involving AKT [
157]. Acadesine (AICAR) is a cell-permeable nucleoside analog that can be metabolically transformed to AICA ribotide (ZMP) by the cells. AICAR can mimic a low-energy state and can regulate the anticancer effects via different mechanisms [
242,
245,
246,
247,
248]. AICAR has been shown to induce selective cell apoptosis, cell proliferation inhibition, and cell cycle arrest in several hematological malignancies [
159,
226,
227,
228,
229] (
Table 2). AICAR showed an acceptable safety profile in a phase I/II clinical trial involving patients with R/R chronic lymphocytic leukemia [
249]. In MCL patients, AICAR showed a cytotoxic effect when combined with rituximab [
159]. AICAR triggers the activation of the AMPK pathway, inhibits the downstream mTOR cascade, and forces MCL cells to enter caspase-dependent apoptosis. The selective BH3-mimetic agent ABT-199 targeting BCL2 increases the sensitivity to AICAR in BCL2-overexpressing MCL cells [
159,
225] (
Table 2,
Figure 1).
2. Potential Metabolic Biomarkers of Lymphoma
Metabolomics has already been used to investigate the pathogenesis of diseases and explore new biomarkers for disease diagnosis, treatments, and prognosis. The PI3K/AKT/mTOR signaling pathway, for instance, has been shown to participate in the cell pro-survival and metabolic reprogramming involving fatty acid metabolism, glycolysis, and tricarboxylic acid cycle in B-cell lymphoma [
250]. Wang et al. generated a metabolic gene panel, using 13 metabolism-associated genes (MAGs) in DLBCL-related metabolic pathways, which divided patients with DLBCL into one of two risk groups. The combination of metabolic gene signatures and other prognostic factors showed a superior prognostic power than IPI and other standard prognostic clinical variables [
250]. Based on the expression patterns of 92 prognosis associated MAGs mining from public database, He et al. divided DLBCL patients into two metabolic clusters with significantly different prognoses [
251]. A prognostic risk model was constructed based on 14 genes selected from 92 MAGs, and then the DLBCL patients were classified into two risk groups, which was superior to the IPI [
251]. These nomograms would be useful in clinical practice and future clinical trials. Serum samples from 100 DLBCL patients and 100 matched healthy controls were analyzed by an untargeted mass-spectrometry-based metabolomics platform. The results of this combined study indicate that 2-AG might play a potential role in the pathogenesis and progression in patients with DLBCL [
252]. The differences in serum metabolites between BLs and normal mice were analyzed by nuclear magnetic resonance-based metabolomics. Glutamate, glycerol, and choline had a high accuracy of diagnosis, which might provide non-invasive approaches for the diagnosis and prognosis of patients with BL [
253]. Serum metabolomics analysis can identify high-risk DLBCL patients with failure of immunochemotherapy, which might provide another novel, non-invasive way for the diagnosis and prognosis of lymphoma [
254]. A study on the skin and plasma of CTCL mice indicated that aberrant metabolites and metabolic pathways were essential metabolic features of CTCLs, whereas accumulative cytidine-5′-triphosphate in adjacent non-involved skin tissues led to CTCL further development [
255]. Changes in lipid profiles provide new biological insights into how MYC regulates cellular metabolism in MYC-induced lymphoma [
256] (
Table 3).
This entry is adapted from the peer-reviewed paper 10.3390/ijms24065493