Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Wei Boon Yap | -- | 2266 | 2023-04-04 16:52:28 | | | |
| 2 | Rita Xu | Meta information modification | 2266 | 2023-04-06 03:25:07 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Mohd Zawawi, Z.; Kalyanasundram, J.; Mohd Zain, R.; Thayan, R.; Basri, D.F.; Yap, W.B. Tumor Necrosis Factor-Alpha (TNF-α) in COVID-19. Encyclopedia. Available online: https://encyclopedia.pub/entry/42793 (accessed on 10 August 2026).
Mohd Zawawi Z, Kalyanasundram J, Mohd Zain R, Thayan R, Basri DF, Yap WB. Tumor Necrosis Factor-Alpha (TNF-α) in COVID-19. Encyclopedia. Available at: https://encyclopedia.pub/entry/42793. Accessed August 10, 2026.
Mohd Zawawi, Zarina, Jeevanathan Kalyanasundram, Rozainanee Mohd Zain, Ravindran Thayan, Dayang Fredalina Basri, Wei Boon Yap. "Tumor Necrosis Factor-Alpha (TNF-α) in COVID-19" Encyclopedia, https://encyclopedia.pub/entry/42793 (accessed August 10, 2026).
Mohd Zawawi, Z., Kalyanasundram, J., Mohd Zain, R., Thayan, R., Basri, D.F., & Yap, W.B. (2023, April 04). Tumor Necrosis Factor-Alpha (TNF-α) in COVID-19. In Encyclopedia. https://encyclopedia.pub/entry/42793
Mohd Zawawi, Zarina, et al. "Tumor Necrosis Factor-Alpha (TNF-α) in COVID-19." Encyclopedia. Web. 04 April, 2023.
Copy Citation
The coronavirus disease 2019 (COVID-19) became a worldwide concern at the beginning of 2020 and has affected millions. High levels of proinflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) and interleukins are produced readily by innate immune cells to fight Severe Acute Respiratory Syndrome-Coronavirus-2 (SARS-CoV-2) infections. The presented work describes the potential of TNF-α in the prognosis, therapeutic and management of COVID-19.
cytokines
COVID-19
inflammation
SARS-CoV-2
TNF-a
1. Introduction
The Severe Acute Respiratory Syndrome-Coronavirus-2 (SARS-CoV-2) is a new beta-coronavirus that causes coronavirus disease 2019 (COVID-19) [1][2][3]. Generally, the infection outcomes vary from person to person and can range from symptomless or mild symptoms to severe respiratory symptoms and failure of multiple vital organs [4][5][6]. One of the severe forms of COVID-19 is acute respiratory distress syndrome (ARDS), which has been reported to be closely associated with the host’s innate immune response [7][8].
ARDS was reported to occur in nearly 16% of hospitalized COVID-19 patients with severe pneumonia [9]. Several proinflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin (IL)-6, IL-2, IL-7, and IL-10 are involved in the development of cytokine release syndrome (CRS), which then contributes to the high morbidity and mortality of COVID-19 including ARDS [10][11][12]. In view of the active involvement of proinflammatory cytokines in the progression of COVID-19, they have been proposed to be part of the molecular targets for diagnosing, treating, and preventing the severe forms of COVID-19 [13]. Therefore, understanding the underlying mechanisms of SARS-CoV-2-induced cytokine storm is particularly critical for patient management and the development of effective treatment regimens.
Upon invasion of viral contagions, tumor necrosis factor-alpha (TNF-α) produced by macrophages and monocytes is one of the early effectors that alert the host’s immunity about the dangers. By binding to the compatible TNF receptor (TNFR1), TNF-α can subsequently induce cellular apoptosis, modulate innate immune responses to limit the replication of the infectious agents, and promote the infiltration of macrophages, dendritic cells, natural killer cells, and neutrophils to the affected area to control and clear the infections [14]. TNFR1 is found in almost all types of cells. Therefore, it can exert various modulating effects on a broad range of cells [15]. TNF-α is also a powerful inducer of nuclear factor kappa B (NF-κB) that is responsible for the expression of multiple proinflammatory genes in the cell nucleus [16]. However, excessive TNF-α production over an extended period can backfire on the host [17]. In rheumatoid arthritis (RA) patients, continuous release of TNF-α in the joint space can sustain tissue inflammation and, thus, leads to bone and cartilage impairment [18]. The use of TNF-α inhibitors has proven beneficial for RA management [19]. In addition, TNF-α blockers are also used to treat glucose tolerance and insulin sensitivity impairment seen in Type-2 diabetes mellitus [20]. In terms of viral diseases such as hepatitis C virus (HCV) infections, anti-TNF-α agents used to prevent overreactive TNF-α-related biological activities are proven safe and beneficial in delaying HCV reactivation that may lead to fatal liver failure. The anti-TNF-α therapy aims to control the active role of TNF-α in promoting liver inflammation and fibrosis in HCV-infected patients [21]. In SARS-CoV infections, it is worth noting that TNF-α-induced inflammation can increase the pathogenesis and preassembly of infective virus particles [22]; therefore, TNF-α inhibitors are recommended to reduce severe disease outcomes.
2. Inflammation in SARS-CoV-2 Infection
2.1. Activation of Innate Immune Response by SARS-CoV-2
Upon SARS-CoV-2 infections, the innate immune response is armed, which is then actively involved in synthesizing proinflammatory cytokines, such as interferons (IFN) and chemokines [23][24]. The IFN-mediated antiviral defense is summarized in Figure 1.
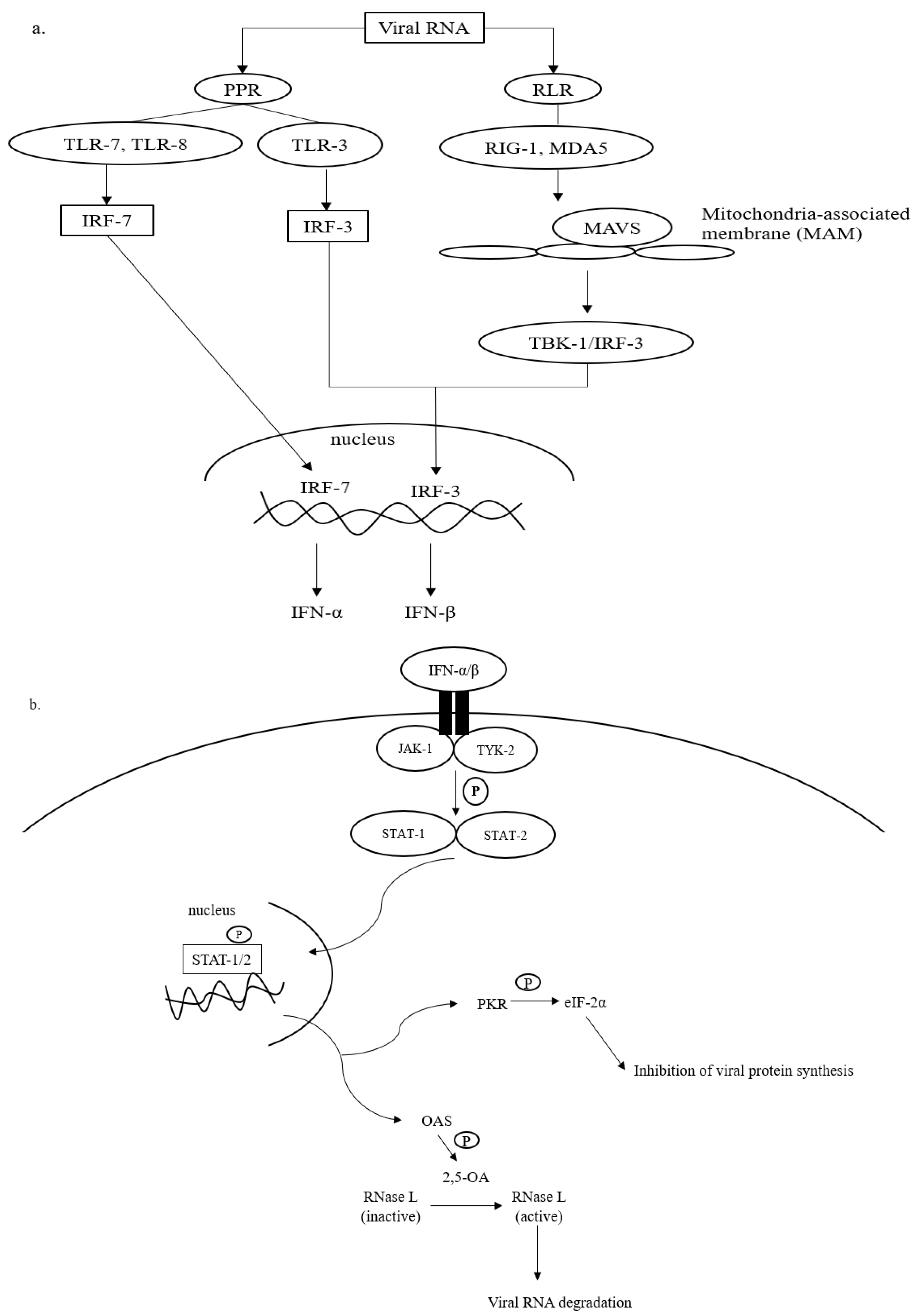
Figure 1. Antiviral defense of IFN. (a) Viral RNA (vRNA) can be recognized by pathogen pattern receptors (PPR) on the cell surface and retinoic acid-like receptors (RLR) in the cytosol. The vRNA sensing by PPR can be initiated by toll-like receptors (TLR)-3, -7, and -8, which then activates gene transcription factors, i.e., interferon regulatory factors (IRF)-3 and -7. IRF-3 and -7 are then translocated into the cell nucleus to express interferon (IFN)-β and interferon-α, respectively. The cytosolic viral dsRNA, on the other hand, is recognized by retinoic-like receptors (RLR) such as retinoic acid-inducible gene-1 (RIG-I) and melanoma differentiation-associated protein 5 (MDA-5). The RLR/vRNA complex subsequently binds to the mitochondrial-antiviral signaling (MAVS) protein located on the mitochondrial outer membrane. Upon the activation of MAVS, IRF-3 is phosphorylated via TANK-binding kinase (TBK)-1 to enhance the expression of IFN-β. (b) The antiviral cascades of IFN α/β demand the activation of Janus kinase/tyrosine kinase/signal transducer and activator of transcription (JAK/TYK/STAT) signaling. Activating the JAK/TYK/STAT transcription signaling pathway results in the expression of antiviral proteins, oligoadenylate synthetase (OAS), and protein kinase R (PKR), which are responsible for viral RNA degradation and inhibition of viral protein synthesis, respectively.
Recognition of intruding viral pathogens by innate immune cells can be initiated via PPRs located on the cell surface, such as Toll-like receptor (TLR)-3, TLR-7, and TLR-8 [25][26]. TLR-3 recognizes viral double-stranded (ds) RNA during the initial phase of infections, whereas TLR-7 and TLR-8 that are found abundantly on bronchial epithelial cells and alveolar macrophages sense the presence of viral single-stranded (ss) RNA prior to the activation of the innate immune cascades (Figure 1a). Sensing vRNA by PPRs leads to the activation of gene transcription factors, namely interferon-regulatory factors (IRF)-3 and -7. IRF-3 and -7 are translocated into the cell nucleus and bind to their complementary promoters to induce the transcription of genes responsible for the synthesis of IFN-α and IFN-β.
In addition to PPRs, vRNA, especially dsRNA, can be detected by retinoic acid-like receptors (RLRs) in the cytosol (Figure 1a). RLRs that are often involved in the recognition of viral dsRNA include retinoid inducible gene 1 (RIG-I) and melanoma differentiation-associated gene 5 (MDA-5). The binding of dsRNA to RIG-1 or MDA-5 complexes that later interact with mitochondria-antiviral signaling (MAVS) protein on the outer membrane of mitochondria. The interaction promotes the phosphorylation of IRF-3 by TANK-binding kinase (TBK). As mentioned earlier, IRF-3 is translocated into the cell nucleus to initiate mainly the expression of IFN-β [25].
To amplify the antiviral response, the expressed IFN-α and -β bind to the compatible receptors, which subsequently initiates the phosphorylation of Janus (JAK) and tyrosine (TYK) kinases (Figure 1b). This leads to the activation of signal transducer and activator of transcription (STAT), particularly STAT-1 and -2. The activated STAT-1 and -2 are important transducers for expressing interferon-stimulated genes (ISGs) in the infected host cell [27]. The ISGs are then translated into antiviral molecules of innate immune response that actively battle virus infections, including oligoadenylate synthetase (OAS) and protein kinase R (PKR). OAS mediates the formation of 2,5-oligoadenylate (2,5-A) via ATP. In the presence of 2-5A, dimerization and activation of RNase L occurs in the cytosol, and the activated RNase L degrades vRNA to prevent viral genome replication and transcription (Figure 1b). PKR is responsible for the phosphorylation of eukaryotic initiation factor (eIF)-2α. The activated eIF-2α terminates viral protein translation [28]. In addition to the direct virucidal effects, the activation of OAS/RNAse L and/or PKR/eIF-2α mechanisms also promotes cellular stress, inflammation, and apoptosis, which in turn causes cell death. Collectively, both antiviral actions prevent further virus replication and spreading in the host [29].
Nonetheless, the perpetual virus infection increases the magnitude of host inflammatory events, resulting in more severe disease outcomes such as cytokine storms, vascular disruption, tissue damage, and organ failure [30]. In addition, to evade the host antiviral defense, viruses have evolved to form counteracting proteins that can interfere with the PPR recognition or the activation of innate immunity. Such a phenomenon is also observed in SARS-CoV-2 infections. The virus produces various structural and nonstructural proteins that can hijack the host’s innate response, suppressing type-I IFN production [31][32]. The inhibition can later affect the expression of antiviral molecules, such as PKR, that are responsible for downregulating the expression of essential viral proteins. Coronaviruses also interrupt the phosphorylation of eIF-2α to further impede the PKR-mediated antiviral response, especially in controlling the cellular translation machinery [33]. For example, the MERS-CoV Open Reading Frame (ORF)4a protein inhibits the binding of PKR to the viral dsRNA. The inhibition reduces the sensitivity of PKR in response to viral dsRNA and thus prevents the translational inhibition of viral proteins [34]. In regards to the inhibition of eukaryotic initiation factors, Xiao et al. (2008) found that at the later stages of SARS-CoV-1 infection, the viral S protein interacted with eIF-3F and subsequently permitted the overexpression of IL-6 and IL-8 that are known to promote an inflammatory response in acute virus infections [35]. Of note, the aforementioned modulations on the host antiviral response initially aim to sustain virus infections, including the novel SARS-CoV-2. However, the resultant increase of virus progeny invariably intensifies the activation of the host’s innate immunity and inflammatory response that eventually leads to hyperinflammation and severe disease complications in patients.
Besides inhibiting the biological activities of IFNs, SARS-CoV-2 also prevents cellular apoptosis to avoid elimination by the host immunity and concomitantly buy more time and space for its replication. Amongst the viral antigens of SARS-CoV-2, ORF3a shows anti-apoptotic effects by lowering the expression levels of apoptotic factors in host cells [36]. In another study that investigated the link between SARS-CoV-2 ORF3a and COVID-19 mortality among 20,000 patients from 23 different countries, the results indicated that ORF3a proteins of the virus isolates derived from countries with high infections and mortality cases were actively mutated [37]. The mutations explained the relatively high virus pathogenesis, disease morbidity, and mortality in those countries. Therefore, in conjunction with the other inhibitory actions on the host immunity, the virus can sustain its replication in hosts and cause the occurrence of severe COVID-19.
Despite the crucial roles of IFN and antiviral molecules in fighting virus infections, they often induce systemic inflammation that is highly possible to result in severe disease outcomes [38]. Plus, to date, several lines of evidence have linked the progression of severe forms of COVID-19 to CRS or cytokine storm syndrome (CSS) caused by the overproduction of proinflammatory cytokines and uncontrollable systemic inflammation [12][39][40]. For instance, Chen et al. (2021) reported that COVID-19 casualties exhibited greater levels of proinflammatory interleukins (IL-2, -6, -8, and -10) and TNF-α compared to the COVID-19 survivors [41]. A similar observation was also documented by Udomsinprasert et al. (2021), in which the circulating and locally produced IL-6, IL-10, and TNF-α were significantly increased in deceased COVID-19 patients [42]. These data imply that proinflammatory cytokines are among the main contributing factors causing severe forms of COVID-19, which are often linked to vital organ failure and death among patients [43].
Considering the more pronounced production of proinflammatory cytokines in deceased COVID-19 patients than in survivors, proinflammatory cytokines are deduced as a strong and independent predictor of patient survival [44][45]. Among the proinflammatory cytokines, the level of TNF-α is consistently higher in severe COVID-19 patients and casualties. Mortaz et al. (2021) investigated the serum level of soluble TNF-α receptors in healthy and COVID-19 patients [46]. The results indicated that the levels of TNF-α receptors were significantly higher in ICU and non-ICU COVID-19 patients than in healthy subjects. The observations reflect the feasibility of using TNF-α as a promising molecular predictor of severe COVID-19 in hospitalized patients.
The exact underlying impacts of SARS-CoV-2-induced hyperinflammation on severe COVID-19 patients are yet to be fully understood. However, the relatively high level of TNF-α in patients at the time of admission is strongly believed to be a contributing factor to irreversible organ damage and life-threatening complications [45][47]. Regardless of the active role of TNF-α in promoting the host’s innate immunity, a few reports found less significant implications of TNF-α in the morbidity and mortality of COVID-19 [48]. As a result, it remains relevant to further define, justify, and delineate the importance of TNF-α in the morbidity and mortality of COVID-19 due to the mixed and inconclusive clinical observations and laboratory analyses.
2.2. Activation of TNF-α Signaling Pathway by SARS-CoV-2 and Its Potential in Virus Containment
Practically, in the presence of dangers, for instance, viruses, the production of TNF-α occurs almost instantly upon stimulation via PPRs [49][50]. TNF-α signaling is mediated by two cellular receptors, namely TNF receptor (TNFR)-1 and TNFR-2. Through interactions with TNFR-1 and -2, several intracellular antiviral cascades are awakened in response to the invading infectious agents [50][51]. Interactions between TNF-α and its receptors are summarised in Figure 2.
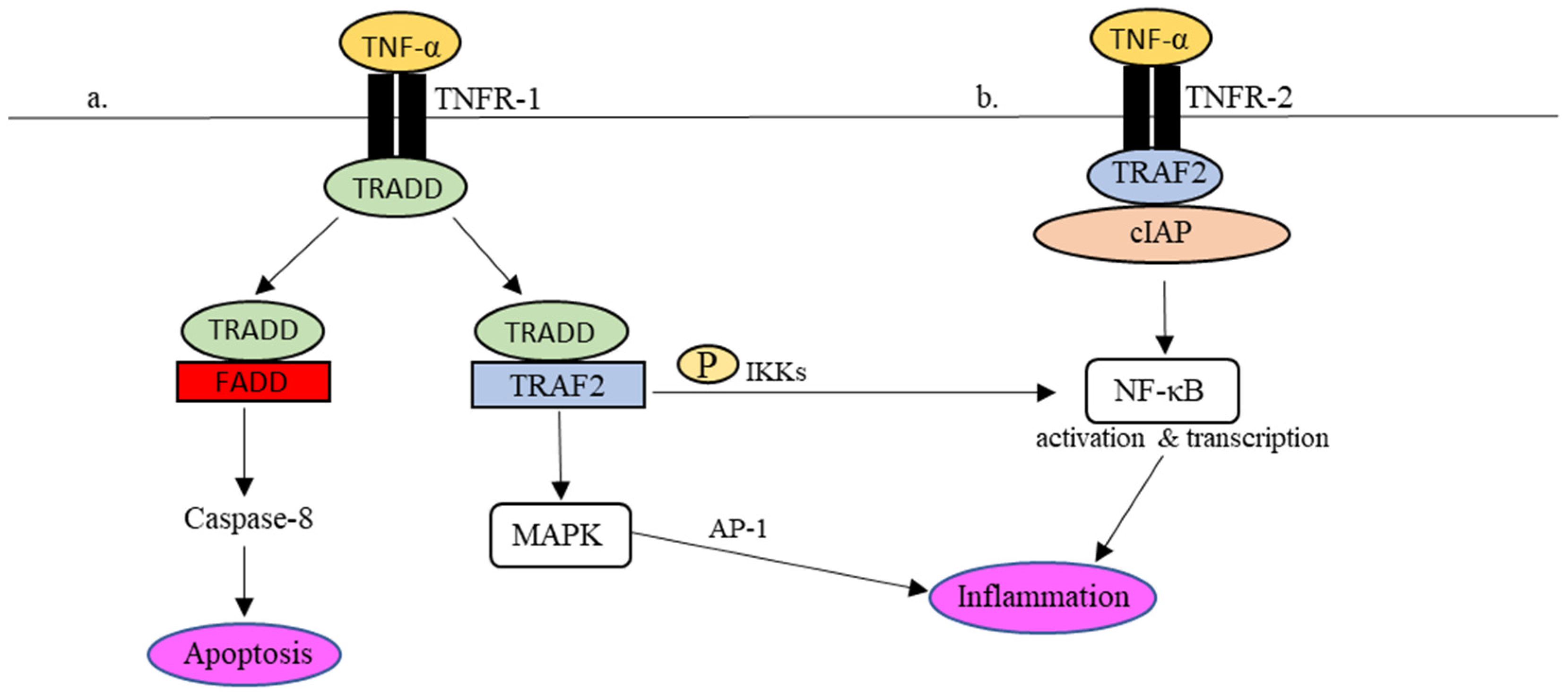
Figure 2. TNFR-1- and TNFR-2-mediated inflammatory responses in the TNF-α signaling pathway. (a) Upon TNFR-1 activation, TRADD is attracted to TNFR1 together with TRAF2 to phosphorylate NF-κβ and activate MAPK as well to regulate the expression of proinflammatory molecules. In addition, TRADD also binds to FADD, which leads to cell apoptosis via caspase-8. (b) In the event of TNFR-2 activation, the recruitment of TRAF2-cIAP complex to TNFR-2 activates the NF-κB pathway, which results in the regulation of inflammation.
Upon binding to TNFR-1 (Figure 2a), the TNFR-1-associated death domain (TRADD) is attracted to the intracellular domain of TNFR-1. The recruitment of TRADD to TNFR-1 further attracts TNF receptors-associated factor 2 (TRAF-2) or Fas-associated protein with death domain (FADD) to control the downstream signaling actions. To activate the transcriptional gene factor, i.e., nuclear factor-kappa beta (NF-κβ), TRAF2 promotes the phosphorylation of NF-κβ by IκB kinases (IKKs). The phosphorylated NF-κβ is active and ready to perform proinflammatory gene transcription in the cell nucleus. In addition, the interaction between TRADD and TRAF-2 also encourages the activation of mitogen-activated protein kinase (MAPK), which in turn stimulates another transcriptional factor, known as activator protein-1 (AP-1), to regulate the expression of proinflammatory molecules, including cytokines and chemokines. As described earlier, uncontrollable cytokine production in response to viral infections may cause acute inflammation [52][53][54]. For example, NF-κβ-mediated IL-6 synthesis in COVID-19 was speculated to be associated with the adversity of the disease [55].
References
- Ludwig, S.; Zarbock, A. Coronaviruses and SARS-CoV-2: A Brief Overview. Anesth. Analg. 2020, 131, 93–96.
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154.
- Mohandas, S.; Yadav, P.D.; Shete, A.; Nyayanit, D.; Sapkal, G.; Lole, K.; Gupta, N. SARS-CoV-2 Delta Variant Pathogenesis and Host Response in Syrian Hamsters. Viruses 2021, 13, 1773.
- Zhou, F.; Yu, T.; Du, R. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062.
- Pascarella, G.; Strumia, A.; Piliego, C.; Bruno, F.; Del Buono, R.; Costa, F.; Scarlata, S.; Agrò, F.E. COVID-19 diagnosis and management: A comprehensive review. J. Intern. Med. 2020, 288, 192–206.
- Rivas, M.N.; Porritt, R.A.; Cheng, M.H.; Bahar, I.; Arditi, M. COVID-19–associated multisystem inflammatory syndrome in children (MIS-C): A novel disease that mimics toxic shock syndrome—The superantigen hypothesis. J. Allergy Clin. Immunol. 2020, 147, 57–59.
- Abdelmoaty, M.M.; Yeapuri, P.; Machhi, J.; Olson, K.E.; Shahjin, F.; Kumar, V.; Pandey, K. Defining the Innate Immune Responses for SARS-CoV-2-Human Macrophage Interactions. Front. Immunol. 2021, 12, 1–15.
- Grant, R.A.; Morales-Nebreda, L.; Markov, N.S.; Swaminathan, S.; Querrey, M.; Guzman, E.R.; Abbott, D.A.; Donnelly, H.K.; Donayre, A.; Goldberg, I.A.; et al. Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature 2021, 590, 635–641.
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Qu, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720.
- Tang, Y.; Liu, J.; Zhang, D.; Xu, Z.; Ji, J.; Wen, C. Cytokine Storm in COVID-19: The Current Evidence and Treatment Strategies. Front. Immunol. 2020, 11, 1708.
- Ragab, D.; Eldin, H.S.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol. 2020, 11, 1446.
- Que, Y.; Hu, C.; Wan, K.; Hu, P.; Wang, R.; Luo, J.; Li, T.; Ping, R.; Hu, Q.; Sun, Y.; et al. Cytokine release syndrome in COVID-19: A major mechanism of morbidity and mortality. Int. Rev. Immunol. 2021, 41, 217–230.
- Reyes, A.; Hu, K.; Teperman, J.; Muskardin, T.W.; Tardif, J.-C.; Shah, B.; Pillinger, M. Anti-inflammatory therapy for COVID-19 infection: The case for colchicine. Ann. Rheum. Dis. 2021, 80, 550–557.
- Jang, D.-I.; Lee, A.-H.; Shin, H.-Y.; Song, H.-R.; Park, J.-H.; Kang, T.-B.; Lee, S.-R.; Yang, S.-H. The Role of Tumor Necrosis Factor Alpha (TNF-α) in Autoimmune Disease and Current TNF-α Inhibitors in Therapeutics. Int. J. Mol. Sci. 2021, 22, 2719.
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M.F. Tumour necrosis factor signalling in health and disease. F1000Research 2019, 8, 111.
- Chen, K.-Y.; Chang, C.-Y.; Hsu, H.-J.; Shih, H.-J.; Huang, I.-T.; Patel, H.H.; Huang, C.-J. Tumor Necrosis Factor-α Mediates Lung Injury in the Early Phase of Endotoxemia. Pharmaceuticals 2022, 15, 287.
- Sinha, P.; Ware, L.B. Selective tumour necrosis factor receptor-1 inhibition in acute lung injury: A new hope or a false dawn? Thorax 2018, 73, 699–701.
- Weaver, A.L. Differentiating the new rheumatoid arthritis biologic thera-pies. JCR J. Clin. Rheumatol. 2003, 9, 99–114.
- Barrera, P.; van Der Maas, A.; Van Ede, A.E.; Kiemeney BA, L.M.; Laan RF, J.M.; Van de Putte LB, A.; van Riel PL, C.M. Drug survival, efficacy and toxicity of monotherapy with a fully human anti-tumour necrosis factor-α anti-body compared with methotrexate in long-standing rheumatoid arthritis. Rheu-Matology 2002, 41, 430–439.
- Borst, S.E. The role of TNF-α in insulin resistance. Endocrine 2004, 23, 177–182.
- Viganò, M.; Degasperi, E.; Aghemo, A.; Lampertico, P.; Colombo, M. Anti-TNF drugs in patients with hepatitis B or C virus infection: Safety and clinical management. Expert Opin. Biol. Ther. 2011, 12, 193–207.
- McDermott, J.E.; Mitchell, H.D.; Gralinski, L.E.; Eisfeld, A.J.; Josset, L.; Bankhead, A.; Neumann, G.; Tilton, S.C.; Schäfer, A.; Li, C.; et al. The effect of inhibition of PP1 and TNFα signaling on pathogenesis of SARS coronavirus. BMC Syst. Biol. 2016, 10, 1–12.
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An introduction to immunology and immunopathology. Allergy Asthma Clin. Immunol. 2018, 14, 1–10.
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.-D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838.
- Park, A.; Iwasaki, A. Type I and type III interferons–induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe 2020, 27, 870–878.
- Ramasamy, S.; Subbian, S. Critical Determinants of Cytokine Storm and Type I Interferon Response in COVID-19 Pathogenesis. Clin. Microbiol. Rev. 2021, 34, e00299-20.
- Amor, S.; Blanco, L.F.; Baker, D. Innate immunity during SARS-CoV-2: Evasion strategies and activation trigger hypoxia and vascular damage. Clin. Exp. Immunol. 2020, 202, 193–209.
- Diamond, M.S.; Kanneganti, T.-D. Innate immunity: The first line of defense against SARS-CoV-2. Nat. Immunol. 2022, 23, 165–176.
- Li, Y.; Renner, D.M.; Comar, C.E.; Whelan, J.N.; Reyes, H.M.; Cardenas-Diaz, F.L.; Weiss, S.R. SARS-CoV-2 induces double-stranded rna-mediated innate immune responses in respiratory epithelial-derived cells and cardiomyocytes. Proc. Natl. Acad. Sci. USA 2021, 118, e2022643118.
- Schultze, J.L.; Aschenbrenner, A.C. COVID-19 and the human innate immune system. Cell 2021, 184, 1671–1692.
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810.
- Lu, Y.; Michel, H.A.; Wang, P.-H.; Smith, G.L. Manipulation of innate immune signaling pathways by SARS-CoV-2 non-structural proteins. Front. Microbiol. 2022, 13, 1027015.
- Cesaro, T.; Michiels, T. Inhibition of PKR by Viruses. Front. Microbiol. 2021, 12, 757238.
- Rabouw, H.H.; Langereis, M.A.; Knaap, R.C.M.; Dalebout, T.J.; Canton, J.; Sola, I.; Enjuanes, L.; Bredenbeek, P.J.; Kikkert, M.; de Groot, R.J.; et al. Middle East Respiratory Coronavirus Accessory Protein 4a Inhibits PKR-Mediated Antiviral Stress Responses. PLoS Pathog. 2016, 12, e1005982.
- Xiao, H.; Xu, L.H.; Yamada, Y.; Liu, D.X. Coronavirus Spike Protein Inhibits Host Cell Translation by Interaction with eIF3f. PLoS ONE 2008, 3, e1494.
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.-Y.; Zhou, W.; Qiu, Y.; et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell. Mol. Immunol. 2020, 17, 881–883.
- Majumdar, P.; Niyogi, S. ORF3a mutation associated with higher mortality rate in SARS-CoV-2 infection. Epide-Miology Infect. 2020, 148, e262.
- Faist, A.; Janowski, J.; Kumar, S.; Hinse, S.; Çalışkan, D.M.; Lange, J.; Ludwig, S.; Brunotte, L. Virus Infection and Systemic Inflammation: Lessons Learnt from COVID-19 and Beyond. Cells 2022, 11, 2198.
- Montazersaheb, S.; Khatibi, S.M.H.; Hejazi, M.S.; Tarhriz, V.; Farjami, A.; Sorbeni, F.G.; Farahzadi, R.; Ghasemnejad, T. COVID-19 infection: An overview on cytokine storm and related interventions. Virol. J. 2022, 19, 1–15.
- Manik, M.; Singh, R.K. Role of toll-like receptors in modulation of cytokine storm signaling in SARS-CoV-2-induced COVID-19. J. Med. Virol. 2021, 94, 869–877.
- Chen, R.; Lan, Z.; Ye, J.; Pang, L.; Liu, Y.; Wu, W.; Qin, X.; Guo, Y.; Zhang, P. Cytokine Storm: The Primary Determinant for the Pathophysiological Evolution of COVID-19 Deterioration. Front. Immunol. 2021, 12, 589095.
- Udomsinprasert, W.; Jittikoon, J.; Sangroongruangsri, S.; Chaikledkaew, U. Circulating Levels of Interleukin-6 and Interleukin-10, But Not Tumor Necrosis Factor-Alpha, as Potential Biomarkers of Severity and Mortality for COVID-19: Systematic Review with Meta-analysis. J. Clin. Immunol. 2020, 41, 11–22.
- Tan, L.Y.; Komarasamy, T.V.; Balasubramaniam, V.R. Hyperinflammatory Immune Response and COVID-19: A Double Edged Sword. Front. Immunol. 2021, 12, 742941.
- Bacci, M.; Leme, R.; Zing, N.P.C.; Murad, N.; Adami, F.; Hinnig, P.; Feder, D.; Chagas, A.; Fonseca, F. IL-6 and TNF-α serum levels are associated with early death in community-acquired pneumonia patients. Braz. J. Med Biol. Res. 2015, 48, 427–432.
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.-H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643.
- Mortaz, E.; Tabarsi, P.; Jamaati, H.; Dalil Roofchayee, N.; Dezfuli, N.K.; Hashemian, S.M.; Moniri, A.; Marjani, M.; Malekmohammad, M.; Mansouri, D.; et al. Increased Serum Levels of Soluble TNF-α Receptor Is Asso-ci-ated With ICU Mortality in COVID-19 Patients. Front Immunol. 2021, 12, 592727.
- Mokhtari, T.; Hassani, F.; Ghaffari, N.; Ebrahimi, B.; Yarahmadi, A.; Hassanzadeh, G. COVID-19 and multiorgan failure: A narrative review on potential mechanisms. Histochem. J. 2020, 51, 613–628.
- Liu, K.; Yang, T.; Peng, X.; Lv, S.; Ye, X.; Zhao, T.; Li, J.; Shao, Z.; Lu, Q.; Li, J.; et al. A systematic meta-analysis of immune signatures in patients with COVID-19. Rev. Med. Virol. 2020, 31, e2195.
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295.
- Yang, S.; Wang, J.; Brand, D.D.; Zheng, S.G. Role of TNF–TNF Receptor 2 Signal in Regulatory T Cells and Its Therapeutic Implications. Front. Immunol. 2018, 9, 784.
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91.
- Darif, D.; Hammi, I.; Kihel, A.; Saik, I.E.I.; Guessous, F.; Akarid, K. The pro-inflammatory cytokines in COVID-19 pathogenesis: What goes wrong? Microb. Pathog. 2021, 153, 104799.
- Wang, W.; Ye, L.; Ye, L.; Li, B.; Gao, B.; Zeng, Y.; Kong, L.; Fang, X.; Zheng, H.; Wu, Z. Up-regulation of IL-6 and TNF-α induced by SARS-coronavirus spike protein in murine macrophages via NF-κB pathway. Virus Res. 2007, 128, 1–8.
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 induces cyto-kine storm with high mortality. Inflamm. Regen. 2020, 40, 1–7.
- Leisman, D.E.; Ronner, L.; Pinotti, R.; Taylor, M.D.; Sinha, P.; Calfee, C.S.; Deutschman, C.S. Cytokine eleva-tion in severe and critical COVID-19: A rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 2020, 8, 1233–1244.
More
Information
Subjects:
Virology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.9K
Entry Collection:
COVID-19
Revisions:
2 times
(View History)
Update Date:
06 Apr 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No