Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sándor Pál | -- | 2297 | 2023-04-01 11:01:00 | | | |
| 2 | Lindsay Dong | + 12 word(s) | 2309 | 2023-04-03 04:44:14 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Akácsos-Szász, O.; Pál, S.; Nyulas, K.; Nemes-Nagy, E.; Fárr, A.; Dénes, L.; Szilveszter, M.; Bán, E.; Tilinca, M.C.; Simon-Szabó, Z. The Pathophysiology of COVID-19 and T2DM Coagulopathy. Encyclopedia. Available online: https://encyclopedia.pub/entry/42709 (accessed on 10 August 2026).
Akácsos-Szász O, Pál S, Nyulas K, Nemes-Nagy E, Fárr A, Dénes L, et al. The Pathophysiology of COVID-19 and T2DM Coagulopathy. Encyclopedia. Available at: https://encyclopedia.pub/entry/42709. Accessed August 10, 2026.
Akácsos-Szász, Orsolya-Zsuzsa, Sándor Pál, Kinga-Ilona Nyulas, Enikő Nemes-Nagy, Ana-Maria Fárr, Lóránd Dénes, Mónika Szilveszter, Erika-Gyöngyi Bán, Mariana Cornelia Tilinca, Zsuzsánna Simon-Szabó. "The Pathophysiology of COVID-19 and T2DM Coagulopathy" Encyclopedia, https://encyclopedia.pub/entry/42709 (accessed August 10, 2026).
Akácsos-Szász, O., Pál, S., Nyulas, K., Nemes-Nagy, E., Fárr, A., Dénes, L., Szilveszter, M., Bán, E., Tilinca, M.C., & Simon-Szabó, Z. (2023, April 01). The Pathophysiology of COVID-19 and T2DM Coagulopathy. In Encyclopedia. https://encyclopedia.pub/entry/42709
Akácsos-Szász, Orsolya-Zsuzsa, et al. "The Pathophysiology of COVID-19 and T2DM Coagulopathy." Encyclopedia. Web. 01 April, 2023.
Copy Citation
Chronic inflammation and endothelium dysfunction are present in diabetic patients. COVID-19 has a high mortality rate in association with diabetes, partially due to the development of thromboembolic events in the context of coronavirus infection. Chronic inflammation, present in DM, enhances the synthesis of several cytokines. This chronic inflammatory state is preceded by a subclinical inflammatory response, represented by elevated IL-1β and IL-6 before the onset of type 2 diabetes mellitus (T2DM). Endothelial dysfunction is also a consequence of Diabetes mellitus (DM) and leads to micro- and macroangiopathy, and concomitantly to hypercoagulability.
COVID-19
thrombosis
coagulopathy
vasculopathy
inflammation
diabetes mellitus
1. The Pathways of Diabetic Endothelial Dysfunction
The most common form of diabetes is type 2 diabetes mellitus (T2DM), a heterogeneous disorder, characterized by relative insulin deficiency, and insulin resistance in target tissues. Insulin resistance could be the key mechanism in the development of T2DM and other pathologies, such as hypertension, obesity, coronary artery disease, and metabolic syndrome [1]. The lack of insulin response is the result of the decrease of insulin receptors on the target cell’s surface. Some authors have reported that altered endothelial cell signaling and activation of redox regulated transcription factors are contributors as well [2][3]. Normally, insulin binding to its receptors activates two major signaling pathways: the phosphatidylinositol 3-kinase (PI3K)-dependent insulin signaling pathway and the mitogen-activated protein kinase (MAPK)-dependent insulin signaling pathway. PI3K is responsible for metabolic changes and is regulating glucose transporter type 4 (GLUT4) translocation in adipose cells, while MAPK regulates mitogenesis, growth, and differentiation [1]. Endothelial production of nitric oxide (NO) is regulated by a PI3K-dependent insulin signaling pathway, with a vasodilator effect, also enhancing glucose uptake of skeletal muscles [1]. It was also described that insulin stimulates endothelin-1 (ET-1) secretion via the MAPK signaling pathway, leading to vasoconstriction. In T2DM, the overproduction of advanced glycation end products (AGEs) and inflammatory cytokines contribute to the development of macroangiopathy, and its main form, atherosclerosis. It was also described that oxidative stress and excess production of reactive oxygen species (ROS) are the consequences of the activated major pathways involved in the development of diabetes- related complications: polyol pathway, protein kinase C (PKC) isoforms, excess formation of AGEs, increased expression of advanced glycation end products (AGEs) receptor and its activating ligands, and overactivity of the hexosamine pathway [4][5][6]. Hyperglycemia in T2DM is also responsible for endothelial dysfunction as the consequence of insulin resistance and excessive production of ROS [1]. Oxidative stress will lead to decreased antioxidant effect and excess synthesis of hydrogen peroxide anion, which directly deactivates NO, resulting in decreased NO activity [4]. ROS in excess can induce epigenetic changes. All these mechanisms can be the common links between the development of diabetes, chronic inflammatory response, and cardiovascular diseases (CVD). Cardiovascular complications are present in approximately 80% of T2DM patients [2].
In T2DM, vascular homeostasis is disturbed by endothelial dysfunction, oxidative stress, platelet hyperreactivity, and inflammation [7], causing alteration in the physicochemical properties of the vascular wall, and enhance the development of atherosclerosis. All these events will aggravate thrombosis and hypercoagulability [8].
2. The Pathomechanism of Endothelial Dysfunction in COVID-19
SARS-CoV-2 infects human cells using the ACE2 receptor and a specific transmembrane serine protease 2 (TMPRSS2), for the priming of the spike protein [9].
ACE2 is expressed in various tissues and organs, including endothelium, lung, heart, intestine, kidney, pancreas, and on the epithelial cells of oral mucosa and the tongue [10]. Reportedly, in T2DM patients the ACE2 receptors are upregulated. It has been hypothesized by many that overexpression of ACE2 receptors in T2DM potentially increases the susceptibility to COVID-19 [11][12].
Once SARS-CoV-2 binds to ACE2 receptors and blocks their activity, the RAAS will be affected. Consequently, accumulation of angiotensin 2 (AngII) will occur, which triggers intracellular signaling pathways (caspase 3, p83 MAPK, ROS, cytochrome C) [13], and leads to vasoconstriction, increased oxidative stress, inflammation, cellular damage, and fibrosis. The regulation of RAAS is influenced by the interaction between ACE2 and bradykinin (BK). Normally, BK acts as a negative regulator of RAAS by dilating blood vessels via local NO release. BK is known for its anti-inflammatory and antioxidant properties and has a role in regulating cytokine production and blood vessel permeability. It also has a stimulating effect on plasminogen secretion and thrombus formation. In COVID-19, the internalization of ACE2 will enhance the activation of different types of BK receptors, leading to increased inflammation and local vascular hyperpermeability. Indirectly, it may activate the coagulation cascade through the resulting endothelial damage [14]. The activation of p83 MAPK can contribute to inflammation and oxidative stress, and the activation of caspase 3 can lead to cellular death [14]. ROS formation will induce oxidative damage to cells and tissues and will release cytochrome C from mitochondria, which can trigger the activation of apoptotic signaling pathways and contribute to cell death as well [15].
Nuclear factor kappa B (NF-κB) is a key molecule involved in the nuclear translocation and activation of controlled genes. Overactivation of NF-κB will lead to the extensive synthesis of proinflammatory mediators, uncontrolled inflammatory response, and eventually to cytochrome storm, as observed in COVID-19 patients [16].
After the endothelial infection by the SARS-CoV-2, von Willebrand factor (vWF) is released into the circulation. The vWF is stored in the Weibel–Palade bodies of the endothelial cells. Platelet aggregation initiated by the increased release of vWF [17] will generate a deposition of platelet-rich clots in the lung microcirculation. This event is the key mechanism leading to respiratory failure [18]. Hypercoagulability will be sustained because of the associated release of factor VIII [17], but it is also the consequence of the virus replication within the endothelial cells. The infection causes endothelial cell death and consequently, the endothelial damage will launch the procoagulant reaction [19].
CAC is characterized by clot formation in the lungs, and elevated D-dimer levels at an early stage of COVID-19, but after the systemic activation of the coagulation and the development of disseminated microthrombosis, multiple organs will be affected [20]. Post-mortem autopsy of severe COVID-19 patients found diffuse alveolar damage, and inflammatory infiltrations with hyaline membrane formation in the lung, but also inflammation of the myocardium, focal pancreatitis, axon injury, glomerular microthrombosis, macrophage accumulation in the brain, and lymphocyte infiltrations of the liver [21].
3. Inflammatory Response in COVID-19
The development of inflammatory processes is a key pathological feature of SARS-CoV-2 infection. From the early beginning of the pandemic, several studies have suggested that massive inflammatory cytokines and chemokines are released in COVID-19 [22].
The innate immune system plays an important role, so proinflammatory cytokine production is a desired phase of the immune response against a pathogen. However, in some cases of COVID-19 infection, proinflammatory cytokine release and synthesis are rapidly overactivated, leading to multisystemic damage to the infected host. Interleukin (IL) 2 and 6 (IL-2, IL-6), tumor necrosis factor-alpha (TNF-α), interferon-gamma (IFN-γ), macrophage inflammatory protein (MIP), and monocyte chemoattractant protein 1 (MCP-1) are among many other cytokines that are present in seriously ill COVID-19 patients [23][24].
3.1. From Cytokine Formation to Cytokine Storm
During inflammation, IL-6-induced tissue factor is released by macrophages [25].
IL-6 is a proinflammatory cytokine that can stimulate the release of other cytokines and activate immune cells, contributing to the overall systemic inflammation observed in severe COVID-19 cases [26].
AngII triggers NF-κB activation, leading to hyperinflammation, mostly through increased synthesis of IL-6 and IL-1b, and subsequently enhancing the transcription of proinflammatory cytokines. These interleukins presented extremely elevated levels in case of severe COVID-19 [27][28].
The exaggerated expression of IL-6 and IL-6 receptor in COVID-19 leads to endothelial cell hyperactivation and a large amount of tissue factor is released, both processes leading to infection-induced coagulopathy. This event plays an important role in thrombocytopenia, although the cytokine storm is the trigger of thrombocytosis. IL-6 is also participating in the production of some coagulation factors (fibrinogen, factor VIII). Acting on the endothelium, IL-6 enhances the synthesis of vascular endothelial growth factor (VEGF), leading to vascular hyperpermeability and hypotension [29]. Additionally, other cytokines, such as TNF-α, IFN-γ, and IL-1β, have also been implicated in the cytokine storm observed in COVID-19 patients and can contribute to increased inflammation and hypercoagulability.
IL-6 and IL-1α have the crucial role of linking inflammation with the coagulation system. During the proinflammatory phase, these cytokines are present on activated platelets, monocytes, and endothelial cells. IL-1α has the role of activating the inflammatory cascade in thrombo-inflammatory conditions, but is also a key element of thrombogenesis, through its granulocyte recruitment effect, prolongation of clot-lysis time, and increasing thrombocyte activity [24]. Combined with TNF, IL-1 is the most important mediator of endogenous coagulation cascade suppression [25].
3.2. Complement Cascade Activation
The activation of the complement system following infection with SARS-CoV-2, as the main participant of innate immunity plays an important role in thrombotic events, combined with endothelium disturbances, thrombocytopenia, and bleeding, all representing risk factors of poor clinical outcome [15].
The literature describes three pathways of complement activation (host–antigen contact, antigen–antibody complex trigger, lectin pathway), all in the defense of the host, leading to the synthesis of C3 and derivatives and activation of plasma proteins [30]. The host–antigen contact will activate the first pathway, the activation of the second pathway is caused by antigen–antibody complexes, and the third one is activated by the lectin pathway, which will bind polysaccharides on antigen surfaces to host cells [31]. At this point, the virus will invade host cells that express the ACE2 receptor and damage them, causing a thrombotic–inflammatory response, which further activates the complement system [32]. The particularity of COVID-19 is related to the lectin pathway component, the mannose-associated serine protease 2 (MASP-2), with a key function of thrombin activation and fibrin mesh formation. Complement cascade participants dysregulate the endothelial cells, affecting the action of clotting cascade proteins [31].
In diabetic patients, the complement system, as an innate humoral defense, will become dysregulated, with the consequences of chronic low-grade inflammation and increased risk of infections [33].
Factor XII (FXIIa) activation has a trigger effect on complement complex C1. A further procoagulant effect of complement activation is the initiation of thrombocyte aggregation [34]. These pathomechanisms reveal a close relationship between complement and coagulation cascade, leading to the reciprocal up-regulation of both processes.
In COVID-19 the complement (C3 and C5) is the mediator for developing inflammation [35]. The terminal C5b-C9 complement activation leads to a release of C3b and C5b fragments, with a proinflammatory role [31]. C3b is involved in the opsonization process, marking the SARS-CoV-2, to be destroyed by immune cells. It is important as well for recruiting macrophages and neutrophils, which can release cytokines, signaling molecules that coordinate the immune response. This will induce prostaglandin and leukotriene synthesis, boosting further proinflammatory cytokine production. In some cases, the release of cytokines can become excessive, leading to an overactive immune response [35].
4. The Cytotoxicity of Neutrophil Extracellular Trap
Neutrophil extracellular trap (NET) release is a mechanism of the innate immune response, as a result of the interaction with activated platelets. It occurs through the explosive intravascular destruction of neutrophils and the release of nucleic substances in the extracellular space, providing a source of extracellular histones with significant cytotoxicity [36]. With the ability to trigger inflammation and thrombosis, NETs release into the extracellular space oxidizes enzymes (NADPH oxidase, nitric oxide synthase) [25][37].
It has also been reported that NETs are among the main drivers of immune-thrombosis in severe COVID-19 cases [38]. Some authors hypothesize that SARS-CoV-2 can directly activate platelets through interaction with its surface spike protein [39], which triggers the release of platelet granules containing proinflammatory and procoagulant factors. Additionally, cytokine release as the result of the immune response to infected cells can also contribute to platelet activation. In COVID-19, platelets are activated and play a role in microvascular thrombosis, leading to serious complications such as acute respiratory distress syndrome (ARDS) and multi-organ failure (MOF) [40].
5. Hypercoagulability
The hypercoagulability present in T2DM will be enhanced by SARS-CoV-2′s binding to the ACE2 receptor and the receptor’s internalization will alter ACE2 functionality. Normally the enzyme binds to AngII, transforming it into angiotensin 1-7 (Ang1-7) peptide with anti-inflammatory effects. Ang1-7 binds and activates a MAS-related transmembrane G-protein coupled receptor (MRGPCR) [15], this reaction will assure anti-inflammatory, antioxidant, and antithrombotic effects. The downregulation of ACE2 receptors will alter RAAS leading to the above-mentioned hypercoagulability, but also hyper-inflammation, hypertension, hypertrophy, and apoptosis [14].
Moreover, platelet dysfunction can also lead to hypercoagulable states [30]. Platelet activation occurs through the initiation of angiotensin II type 1 receptor (AT1R) and its release of plasminogen activator inhibitor 1 (PAI-1). Platelets are also triggered by the altered ACE2R function [41]. Another important aspect is that platelets have MRGPCRs that modify thrombosis via NO release, and this also contributes to clot formation. This may be the explanation for the importance of platelet activation in COVID-19 coagulopathy [41].
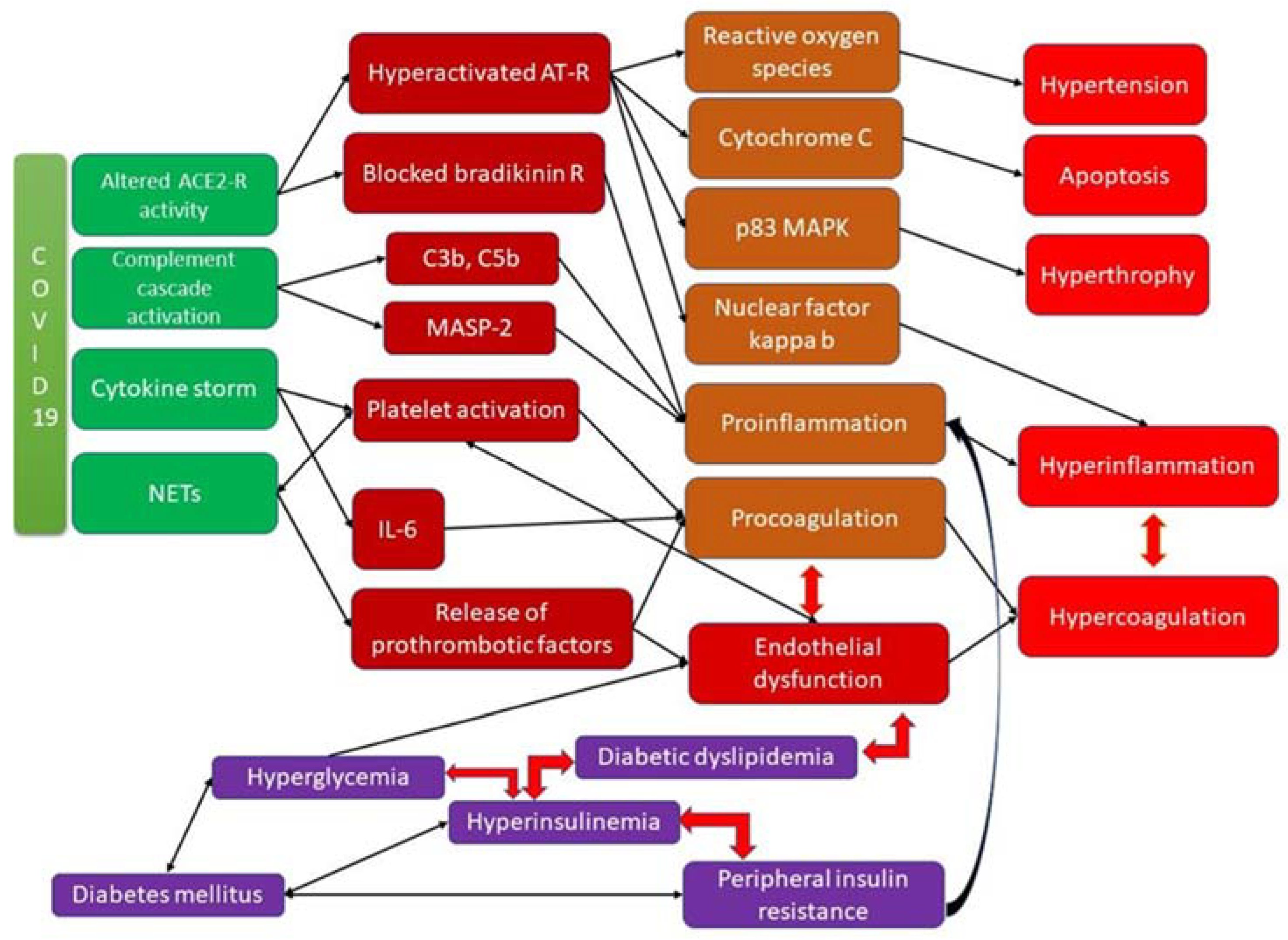
The interrelation between different factors and pathways involved in the development of arteriopathy and coagulopathy in diabetic patients is presented in Figure 1.
Figure 1. Coagulopathy in DM and COVID-19 infection and the underlying molecular mechanisms.
6. The Importance of Genetic Background
The novel coronavirus breakout and pandemic have intensified the need for genetic investigations related to gene expression for a better understanding of the underlying pathomechanisms of SARS-CoV-2 and its genetic association with different diseases [42][43].
SARS-CoV-2 is a coronavirus of bat origin, causing a disease with various symptoms, from mild fever, cough, and sore throat in some patients or severe pneumonia, ARDS, and even septic shock or MOF in other individuals [44].
The intracellular pathogenicity of viruses makes them dependent on host cells, but it also suggests a virus–host protein–protein interaction (PPI). These PPIs have been the focus of recent analyses. The identification of the most common human proteins known to interact with coronavirus could provide a better understanding of the mechanism of COVID-19 and may suggest therapeutic strategies or drug combinations [45][46].
Using a network-based strategy, which incorporates gene expression profiling, gene ontologies, and PPI analysis, RNA-Seq scientists can identify molecular interactions between virus and host during the development of the infection and establish adequate treatment methods. RNA-Seq is a next-generation sequencing technology to measure gene expression with a high level of accuracy [46].
References
- Muniyappa, R.; Chen, H.; Montagnani, M.; Sherman, A.; Quon, M.J. Endothelial dysfunction due to selective insulin resistance in vascular endothelium: Insights from mechanistic modeling. Am. J. Physio. Endocrinol. Metab. 2020, 319, E629–E646.
- Munteanu, C.; Rotariu, M.; Turnea, M.A.; Anghelescu, A.; Albadi, I.; Dogaru, G.; Silișteanu, S.C.; Ionescu, E.V.; Firan, F.C.; Ionescu, A.M.; et al. Topical Reappraisal of Molecular Pharmacological Approaches to Endothelial Dysfunction in Diabetes Mellitus Angiopathy. Curr. Issues Mol. Biol. 2022, 44, 3378–3397.
- Love, M.K.; Barrett, E.J.; Malin, S.K.; Reusch, J.E.B.; Regensteiner, J.G.; Liu, Z. Diabetes pathogenesis and management: The endothelium comes of age, J. Mol. Cell Biol. 2021, 13, 500–512.
- Maruhashi, T.; Higashi, Y. Pathophysiological Association between Diabetes Mellitus and Endothelial Dysfunction. Antioxidants 2021, 10, 1306.
- Jung, C.H.; Mok, J.O. Recent updates on vascular complications in patients with type 2 diabetes mellitus. Endocrinol. Metab. 2020, 35, 260–271.
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275.
- Iacobini, C.; Vitale, M.; Pesce, C.; Pugliese, G.; Menini, S. Diabetic Complications and Oxidative Stress: A 20-Year Voyage Back in Time and Back to the Future. Antioxidants 2021, 10, 727.
- Gusev, E.; Sarapultsev, A.; Hu, D.; Chereshnev, V. Problems of Pathogenesis and Pathogenetic Therapy of COVID-19 from the Perspective of the General Theory of Pathological Systems (General Pathological Processes). Int. J. Mol. Sci. 2021, 22, 7582.
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8.
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12, 8.
- Pinchera, B.; Scotto, R.; Buonomo, A.; Zappulo, E.; Stagnaro, F.; Gallicchio, A.; Viceconte, G.; Sardanelli, A.; Mercinelli, S.; Villari, R. Diabetes and COVID-19: The potential role of mTOR. Diabetes Res. Clin. Pract. 2022, 186, 109813.
- Calvisi, S.L.; Ramirez, G.A.; Scavini, M.; Da Prat, V.; di Lucca, G.; Laurenzi, A.; Gallina, G.; Cavallo, L.; Borio, G.; Farolfi, F.; et al. Thromboembolism risk among patients with diabetes/stress hyperglycemia and COVID-19. Metabolism 2021, 123, 154845.
- Lumbers, E.R.; Delforce, S.J.; Pringle, K.; Smith, G.R. The Lung, the Heart, the Novel Coronavirus, and the Renin-Angiotensin System; The Need for Clinical Trials. Front. Med. (Lausanne) 2020, 22, 248.
- Scialo, F.; Daniele, A.; Amato, F.; Pastore, L.; Matera, M.G.; Cazzola, M.; Castaldo, G.; Bianco, A. ACE2: The Major Cell Entry Receptor for SARS-CoV-2. Lung 2020, 198, 867–877.
- Ali, M.A.; Spinler, S.A. COVID-19 and thrombosis: From bench to bedside. Trends Cardiovasc. Med. 2021, 31, 143–160.
- Gudowska-Sawczuk, M.; Mroczko, B. The Role of Nuclear Factor Kappa B (NF-κB) in Development and Treatment of COVID-19: Review. Int. J. Mol. Sci. 2022, 23, 5283.
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.-H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582.
- Iba, T.; Levy, J.H.; Levi, M.; Connors, J.M.; Thachil, J. Coagulopathy of Coronavirus Disease 2019. Crit. Care Med. 2020, 48, 1358–1364.
- Labò, N.; Ohnuki, H.; Tosato, G. Vasculopathy and Coagulopathy Associated with SARS-CoV-2 Infection. Cells 2020, 9, 1583.
- Iba, T.; Warkentin, T.E.; Thachil, J.; Levi, M.; Levy, J.H. Proposal of the Definition for COVID-19-Associated Coagulopathy. J. Clin. Med. 2021, 10, 191.
- Eketunde, A.O.; Mellacheruvu, S.P.; Oreoluwa, P. A review of postmortem findings in patients with COVID-19. Cureus 2020, 12, e9438.
- Gemmati, D.; Bramanti, B.; Serino, M.L.; Secchiero, P.; Zauli, G.; Tisato, V. COVID-19 and Individual Genetic Susceptibility/Receptivity: Role of ACE1/ACE2 Genes, Immunity, Inflammation and Coagulation. Might the Double X-chromosome in Females Be Protective against SARS-CoV-2 Compared to the Single X-Chromosome in Males? Int. J. Mol. Sci. 2020, 21, 3474.
- Lazzaroni, M.G.; Piantoni, S.; Masneri, S.; Garrafa, E.; Martini, G.; Tincani, A.; Andreoli, L.; Franceschini, F. Coagulation dysfunction in COVID-19: The interplay between inflammation, viral infection and the coagulation system. Blood Rev. 2021, 46, 100745.
- Savla, S.R.; Prabhavalkar, K.S.; Bhatt, L.K. Cytokine storm associated coagulation complications in COVID-19 patients: Pathogenesis and Management. Expert Rev. Anti-Infect Ther. 2021, 19, 1397–1413.
- Gómez-Mesa, J.E.; Galindo-Coral, S.; Montes, M.C.; Martin, A.J.M. Thrombosis and Coagulopathy in COVID-19. Curr. Probl. Cardiol. 2021, 46, 100742.
- Ragab, D.; Salah Eldin, H.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol. 2020, 11, 1446.
- Mascolo, A.; Scavone, C.; Rafaniello, C.; Ferrajolo, C.; Racagni, G.; Berrino, L.; Paolisso, G.; Rossi, F.; Capuano, A. Renin-Angiotensin System and Coronavirus Disease 2019: A Narrative Review. Front. Cardiovasc. Med. 2020, 7, 143.
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590.
- Tomerak, S.; Khan, S.; Almasri, M.; Hussein, R.; Abdelati, A.; Aly, A.; Salameh, M.A.; Saed Aldien, A.; Naveed, H.; Elshazly, M.B.; et al. Systemic inflammation in COVID-19 patients may induce various types of venous and arterial thrombosis: A systematic review. Scand J. Immunol. 2021, 94, e13097.
- Kaiafa, G.; Savopoulos, C.; Karlafti, E.; Pantazi, K.; Paramythiotis, D.; Thomaidou, E.; Daios, S.; Ztriva, E.; Gionis, M.; Fyntanidou, V.; et al. Coagulation Profile of COVID-19 Patients. Life 2022, 12, 1658.
- Tomo, S.; Kumar, K.P.; Roy, D.; Sankanagoudar, S.; Purohit, P.; Yadav, D.; Banerjee, M.; Sharma, P.; Misra, S. Complement activation and coagulopathy—An ominous duo in COVID19. Expert Rev. Hematol. 2021, 14, 155–173.
- Conway, E.M.; Pryzdial, E.L.G. Is the COVID-19 thrombotic catastrophe complement-connected? J. Thromb. Haemost. 2020, 18, 2812–2822.
- Pérez-Galarza, J.; Prócel, C.; Cañadas, C.; Aguirre, D.; Pibaque, R.; Bedón, R.; Sempértegui, F.; Drexhage, H.; Baldeón, L. Immune Response to SARS-CoV-2 Infection in Obesity and T2D: Literature Review. Vaccines 2021, 9, 102.
- Hollenberg, M.D.; Epstein, M. The innate immune response, microenvironment proteinases, and the COVID-19 pandemic: Pathophysiologic mechanisms and emerging therapeutic targets. Kidney Int. Suppl. 2022, 12, 48–62.
- Smail, S.W.; Saeed, M.; Alkasalias, T.; Khudhur, Z.O.; Younus, D.A.; Rajab, M.F.; Abdulahad, W.H.; Hussain, H.I.; Niaz, K.; Safdar, M. Inflammation, immunity and potential target therapy of SARS-COV-2: A total scale analysis review. Food Chem. Toxicol. 2021, 150, 112087.
- Zuo, Y.; Zuo, M.; Yalavarthi, S.; Gockman, K.; Madison, J.A.; Shi, H.; Woodard, W.; Lezak, S.P.; Lugogo, N.L.; Knight, J.S.; et al. Neutrophil extracellular traps and thrombosis in COVID-19. J. Thromb. Thrombolysis 2021, 51, 446–453.
- Manolis, A.S.; Manolis, T.A.; Papatheou, D.; Melita, H. COVID-19 Infection: Viral Macro- and Micro-Vascular Coagulopathy and Thromboembolism/Prophylactic and Therapeutic Management. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 12–24.
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157.
- Shen, S.; Zhang, J.; Fang, Y.; Lu, S.; Wu, J.; Zheng, X.; Deng, F. SARS-CoV-2 interacts with platelets and megakaryocytes via ACE2-independent mechanism. J. Hematol. Oncol. 2021, 14, 72.
- Thierry, A.R.; Roch, B. Neutrophil Extracellular Traps and By-Products Play a Key Role in COVID-19: Pathogenesis, Risk Factors, and Therapy. J. Clin. Med. 2020, 9, 2942.
- Ahmed, S.; Zimba, O.; Gasparyan, A.Y. Thrombosis in Coronavirus disease 2019 (COVID-19) through the prism of Virchow’s triad. Clin. Rheumatol. 2020, 39, 2529–2543.
- Nashiry, A.; Sumi, S.S.; Islam, S.; Quinn, J.M.W.; Moni, A.M.A. Bioinformatics, and system biology approach to identify the influences of COVID-19 on cardiovascular and hypertensive comorbidities. Brief Bioinform. 2021, 22, 1387–1401.
- Islam, M.B.; Chowdhury, U.N.; Nain, Z.; Uddin, S.; Ahmed, M.B.; Moni, M.A. Identifying molecular insight of synergistic complexities for SARS-CoV-2 infection with pre-existing type 2 diabetes. Comput. Biol. Med. 2021, 136, 104668.
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506.
- Liu, C.; Ma, Y.; Zhao, J.; Nussinov, R.; Zhang, Y.C.; Cheng, F.; Zhang, Z.K. Computational network biology: Data, model, and applications. Phys. Rep. 2020, 846, 1–66.
- Khan, A.A.; Khan, Z. Comparative host–pathogen protein–protein interaction analysis of recent coronavirus outbreaks and important host targets identification. Brief Bioinform. 2021, 22, 1206–1214.
More
Information
Subjects:
Infectious Diseases
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Entry Collection:
COVID-19
Revisions:
2 times
(View History)
Update Date:
03 Apr 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No