Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Cristian Sandoval | -- | 2009 | 2023-03-30 06:06:05 | | | |
| 2 | Camila Xu | Meta information modification | 2009 | 2023-03-30 07:10:16 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sandoval, C.; Calle, Y.; Godoy, K.; Farías, J. Pathophysiology of Acute Myeloid Leukemia. Encyclopedia. Available online: https://encyclopedia.pub/entry/42630 (accessed on 23 July 2026).
Sandoval C, Calle Y, Godoy K, Farías J. Pathophysiology of Acute Myeloid Leukemia. Encyclopedia. Available at: https://encyclopedia.pub/entry/42630. Accessed July 23, 2026.
Sandoval, Cristian, Yolanda Calle, Karina Godoy, Jorge Farías. "Pathophysiology of Acute Myeloid Leukemia" Encyclopedia, https://encyclopedia.pub/entry/42630 (accessed July 23, 2026).
Sandoval, C., Calle, Y., Godoy, K., & Farías, J. (2023, March 30). Pathophysiology of Acute Myeloid Leukemia. In Encyclopedia. https://encyclopedia.pub/entry/42630
Sandoval, Cristian, et al. "Pathophysiology of Acute Myeloid Leukemia." Encyclopedia. Web. 30 March, 2023.
Copy Citation
Acute myeloid leukemia is a cancerous condition that affects hemopoietic stem cells or progenitors and is defined by the stopping of myeloid lineage development and abnormal proliferation.
adults
cytochrome
monooxygenase
polymorphisms
1. Introduction
Acute myeloid leukemia is a cancerous condition that affects hemopoietic stem cells or progenitors and is defined by the stopping of myeloid lineage development and abnormal proliferation [1]. The most prevalent acute leukemia in adults is acute myeloid leukemia, which has a wide range of genetic variations. In 2021, more than 20,000 new cases of acute myeloid leukemia were expected in the US [2]. Traditionally, acute myeloid leukemia has been categorized based on immunophenotype and morphology. However, genetic abnormalities, such as chromosomal translocations and transcription factor involvement, must be considered in acute myeloid leukemia diagnostic algorithms [3][4]. These factors led to the classification of acute myeloid leukemia into six groups [3]: myeloid proliferations linked to Down syndrome, myeloid sarcoma, recurring genetic abnormalities, therapy-related myeloid neoplasms, and acute myeloid leukemia with myelodysplasia-related alterations.
2. Pathophysiology of Acute Myeloid Leukemia
2.1. Cytogenetic Abnormalities
Acute myeloid leukemia is characterized by mutations in hematopoiesis-related genes [5]. Ineffective erythropoiesis and bone marrow failure are caused by these mutations, which cause a clonal increase in undifferentiated myeloid progenitors (blasts) in the peripheral blood and bone marrow. Recent research has suggested that it could result from several recurring genetic changes in hematopoietic stem cells that accumulate over time [6][7][8][9][10][11][12]. Acute myeloid leukemia often develops from scratch in a previously healthy person. Although the precise source of genetic abnormalities is unknown, a few known risk factors include smoking, chemotherapy, and radiation exposure [13]. Aplastic anemia, paroxysmal nocturnal hemoglobinuria, myelodysplastic syndrome, and myeloproliferative diseases can all develop into acute myeloid leukemia [14][15].
Genetic mutations that have familial causes should also be considered (Table 1). The most prevalent mutational subset in acute myeloid leukemia is type 1 mutations, which are present in about two-thirds of patients and result in abnormal activation and proliferation of cellular signaling pathways (e.g., FMS-like tyrosine kinase 3 (FLT3); Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS); NRAS Proto-Oncogene, GTPase (NRAS); Tyrosine-protein phosphatase non-receptor type 11 (PTPN11); neurofibromin 1 (NF1); and KIT proto-oncogene, receptor tyrosine kinase (KIT)). It is interesting to note that mutations in this class are usually found in subclonal cellular fractions, indicating that they are frequently late clonal events in the development of illness [6].
Table 1. Recurrent mutations in acute myeloid leukemia.
| Functional Class | Specific Example Mutations | References |
|---|---|---|
| Signaling and kinase pathways | FLT3, KRAS, NRAS, KIT, PTPN11, and NF1 | [6] |
| DNA methylation and chromatin modification | DNMT3A, IDH1, IDH2, TET2, ASXL1, EZH2, and MLL/KMT2A | [6][16][17] |
| Nucleophosmin | NPM1 | [18][19] |
| Transcription factors | CEBPA, RUNX1, and GATA2 | [20][21][22][23][24][25][26][27] |
| Tumor suppressors | TP53 | [28][29][30] |
| Spliceosome complex | SRSF2, U2AF1, SF3B1, and ZRSR2 | [31][32] |
| Cohesin complex | RAD21, STAG1, STAG2, SMC1A, and SMC3 | [33][34] |
Somatic mutations within key epigenetic regulators are identified in >50% of acute myeloid leukemia, and are now recognized as a key, and often an inciting, component of leukemogenesis [6]. It is of interest that age-related clonal hematopoiesis, identified in >10% of individuals over age 65, is predominantly defined by the clonal outgrowth of preleukemic clones harboring mutations in one of the genes within this epigenetic class [16][17].
Along with FLT3 and DNA methyltransferase 3 alpha (DNMT3A; https://www.ncbi.nlm.nih.gov/gene/1788; accessed on 14 January 2023), nucleophosmin 1 (NPM1) is one of the three most frequent driver mutations in acute myeloid leukemia. The regulation of pathways for cell proliferation, differentiation, adhesion, and death by receptor tyrosine kinase (RTKs) signaling pathways in acute myeloid leukemia is crucial for the onset and spread of malignancy. Around 20 separate subfamilies make up the RTKs, which include class III and TYRO3, AXL, and MERTK (TAM) family RTKs [35]. Class III RTKs, which include c-Kit, CSF1R, FLT3, and platelet-derived growth factor receptors (PDGFR), have been discovered to have a major effect on leukemogenesis and transformation into acute myeloid leukemia. Class III RTKs have been linked to aberrant activation that promotes proliferation in leukemia. Particularly, FLT3 expression and c-Kit mutations are critical for acute myeloid leukemia. Both RTKs have been crucial targets in the development of antileukemic therapies, since they are both associated with worse prognoses. TYRO3, AXL, and MERTK are members of the TAM family of RTKs. These are essential for platelet activation and stabilization, for the normal hematopoiesis of certain innate immune cells, and have been linked to erythropoiesis [19]. TAM RTKs are critical for normal hematopoietic development, but they can activate pathways for proliferation and survival in cancer cells, particularly in acute leukemia [36]. TAM RTKs, particularly AXL and MERTK, have been linked to hematologic malignancies and have grown in interest as potential targets for creating novel treatments [36][37][38][39][40].
In addition, compared to the genomes of other cancers, acute myeloid leukemia genomes are typically less mutated [41], with similar distributions of mutations before and following relapse [42][43]. Of these mutations, many frequently occur in genes involved in DNA methylation and epigenetic regulation, such as DNMT3A, TET1/2, and IDH1/2 [44]. Hassan et al. [45] point to a greater need for understanding acute myeloid leukemia through a non-genetic lens, focusing on DNA methylation and other epigenetic modalities. They also suggest relative independence between the progression of acute myeloid leukemia and the disease’s strictly genetic landscape.
IDH1 gene mutations are present in around 6–10% of individuals with acute myeloid leukemia [46]. Given its capacity to create cytoplasmic NADPH and glucose sensing, IDH1 is implicated in controlling cellular metabolism, particularly lipid metabolism [47][48]. Isocitrate is oxidized to α-ketoglutarate by wild-type isocitrate dehydrogenases [49]. It has been hypothesized that the IDH1 Arg132 mutation alters the way the enzyme functions, causing α-ketoglutarate to be converted to R(—)-2-hydroxyglutarate [50]. This excess of R(—)-2-hydroxyglutarate causes cellular proliferation to rise and cellular differentiation to be compromised [51].
Nucleophosmin 1 mutations, occurring almost exclusively within exon 12 of the gene, occur in approximately one-third of adults with acute myeloid leukemia, and in more than 50% of NK-AML. The NPM1 gene encodes for the nuclear chaperone protein NPM, which shuttles between the nucleus and cytoplasm and plays a role in diverse cellular functions, including protein formation, ribosome biogenesis, DNA replication, and the cell cycle. NPM1 mutations are typically stable throughout the disease course, are identified in nearly all leukemic cells, and impart a distinct expression profile [52]. NPM1 mutations in the setting of mutant DNMT3A, particularly in the setting of FLT3 internal tandem duplication (FLT3-ITD), confer a markedly poor prognosis [6][53].
Approximately 20% to 25% of adults with acute myeloid leukemia have mutations involving the myeloid transcription factors Runt-related transcription factor 1 (RUNX1), CEBPA, and/or GATA binding protein 2 (GATA2) [5]. In this sense, RUNX1 is known to act as a direct transcriptional activator of several proteins important for platelet function and as a transcriptional repressor of others, including MYH10 [54][55][56][57] and ANKRD26 [58]. In addition, CEBPA encodes a master hematopoietic transcription factor that acts as a critical regulator of granulocyte and monocyte differentiation [59], while GATA2 encodes a zinc finger transcription factor critical for normal hematopoiesis [60][61] and lymphatic vascular development [62][63].
Tumor protein p53 (TP53) is a key tumor suppressor with highly variable functions related to the maintenance of genomic stability, including regulation of cellular senescence, apoptosis, metabolism, and DNA repair. Although uncommon in de novo acute myeloid leukemia, TP53 mutations occur in ~15% of therapy-related acute myeloid leukemia or acute myeloid leukemia with myelodysplastic syndrome-related changes, and are predominantly associated with complex cytogenetics, advanced age, chemotherapy resistance, and poor survival [29][30]. Irrespective of age or treatment modality, TP53 mutations in acute myeloid leukemia portend lower response rates and inferior outcomes compared with TP53 wild-type acute myeloid leukemia patients [31].
Frequently mutated in myelodysplastic syndrome and myeloproliferative neoplasms, mutations in splicing factors (SF3B1, SRSF2, U2AF1, and ZRSR2) are identified in ~10% of patients with acute myeloid leukemia and are associated with older age, less proliferative disease, poor rates of response to standard treatment, and decreased survival. Spliceosome mutations are postulated to promote malignancy through the missplicing of various genes involved in epigenetic regulation, transcription, and genome integrity [32].
The structural maintenance of chromosomes (SMC3 and SMC1A), RAD cohesin complex component (RAD21), and cohesin subunit SA (STAG1/STAG2) make up the four core elements of cohesin with a ring shape. Cohesin helps several other subunits, such as NIPBL, MAU2, WAPL, PDS5A, PDS5B, and sororin, to form cohesion during the cell cycle [64][65][66][67]. Consequently, the ring-shaped cohesin controls the sister chromatids’ separation, DNA replication, and repair of the broken double-strand DNA during the advancement of the cell cycle [68][69][70][71][72]. To control chromatin structure and gene expression, the cohesin complex can also interact with the transcriptional repressor CTCF, promoters, mediators, enhancers, initiation and elongation forms of RNA polymerase II (RNAPII), or transcription factors (TFs) [69][73][74][75][76][77].
Acute myeloid leukemia prognosis is exceedingly variable and unpredictable. It can be caused by molecular changes, chromosomal translocations, or genetic mutations. Genetic mutations have been shown to occur in around 97% of cases. Table 2 shows an updated classification of acute myeloid leukemia based on the National Comprehensive Cancer Network’s cytogenetic and molecular criteria [78][79]. The following are examples of cytogenetic subsets: (1) chromosomal translocations [t(15;17)(q22,q21)] and core binding factor acute myeloid leukemia (CBF-AML), both of which are cytogenetic/molecular subgroups of inversion 16 [inv16(p13;q22)] or t(16;16)(p13;q22); (2) individuals with cytogenetically normal acute myeloid leukemia (CN-AML) who have monosomy 5 or 7 or t(9 or 11) have a low risk (40–50% of patients) [67][68][69][70][71][72]; (3) individuals with t(6;9), inv (3), or 11q changes (11q23 translocations) [67][68][69][70][71][72]; (4) and those with other karyotypes have been demonstrated to have a higher risk of treatment failure and mortality [80][81][82][83][84][85]. Furthermore, people with translocations involving the MECOM (myelodysplastic syndrome-1 and ecotropic viral integration site 1 (EVI1) complex locus) gene on chromosome 3q26.2 have a very poor prognosis [86][87].
Table 2. Classification of acute myeloid leukemia based on the National Comprehensive Cancer Network’s cytogenetic and molecular criteria.
| Type | Diagnostic Criteria |
|---|---|
| Acute myeloid leukemia with minimal differentiation | Blasts are negative (<3%) for MPO and SBB. |
| Expression of two or more myeloid-associated antigens, such as CD13, CD33, and CD117. | |
| Acute myeloid leukemia without maturation | ≥3% blasts positive for MPO or SBB and negative for NSE. |
| Maturing cells of the granulocytic lineage constitute <10% of the nucleated bone marrow cells. | |
| Expression of two or more myeloid-associated antigens, such as MPO, CD13, CD33, and CD117. | |
| Acute myeloid leukemia with maturation | ≥3% blasts positive for MPO or SBB. |
| Maturing cells of the granulocytic lineage constitute ≥10% of the nucleated bone marrow cells. | |
| Monocyte lineage cells constitute <20% of bone marrow cells. | |
| Expression of two or more myeloid–associated antigens, such as MPO, CD13, CD33, and CD117. | |
| Acute basophilic leukemia | Blasts and immature/mature basophils with metachromasia on toluidine blue staining. |
| Blasts are negative for cytochemical MPO, SBB, and NSE. | |
| No expression of strong CD117 equivalent (to exclude mast cell leukemia). | |
| Acute myelomonocytic leukemia | ≥20% monocytes and their precursors. |
| ≥20% maturing granulocytic cells. | |
| ≥3% of blasts positive for MPO. | |
| Acute monocytic leukemia | ≥80% monocytes and/or their precursors (monoblasts and/or promonocytes). |
| <20% maturing granulocytic cells. | |
| Blasts and promonocytes expressing at least two monocytic markers including CD11c, CD14, CD36 and CD64, or NSE. | |
| Acute erythroid leukemia | ≥30% immature erythroid cells (proerythroblasts) |
| Bone marrow with erythroid predominance, usually ≥80% of cellularity | |
| Acute megakaryoblastic leukemia | Blasts express at least one or more of the platelet glycoproteins: CD41 (glycoprotein llb), CD61 (glycoprotein IIIa), or CD42b (glycoprotein lb) |
MPO: myeloperoxidase; NSE: nonspecific esterase–butyrate; SBB: Sudan Black B.
2.2. Mutations
Modern molecular technologies have permitted the identification of a wide spectrum of genetic disorders. Six genes have already been incorporated into the European Leukemia Net risk categories [88], including FLT3, NPM1, CCAAT/enhancer binding protein α (CEBPA), RUNX1, additional sexcombs-like 1 (ASXL1), and TP53 [88]. Other recurrent gene mutations in acute myeloid leukemia patients have been discovered [89][90][91][92][93][94]. Furthermore, research has been undertaken on the essential roles played by recurrent gene mutations in the pathogenesis of acute myeloid leukemia, as well as the development of medicines that precisely target gene alterations [95][96][97][98][99][100][101]. Preleukemic cell detection, particularly in acute myeloid leukemia patients with mutant DNMT3A and TET2 genes, has been linked to leukemia genesis [102][103]. Mutations in the DNMT3A and TET2 genes are common in patients with clonal hematopoiesis of undetermined potential [16][17][104][105]; these mutations may serve as markers for the identification of preleukemia cells [102][103]. A summary of the pathophysiology of acute myeloid leukemia is shown in Figure 1.
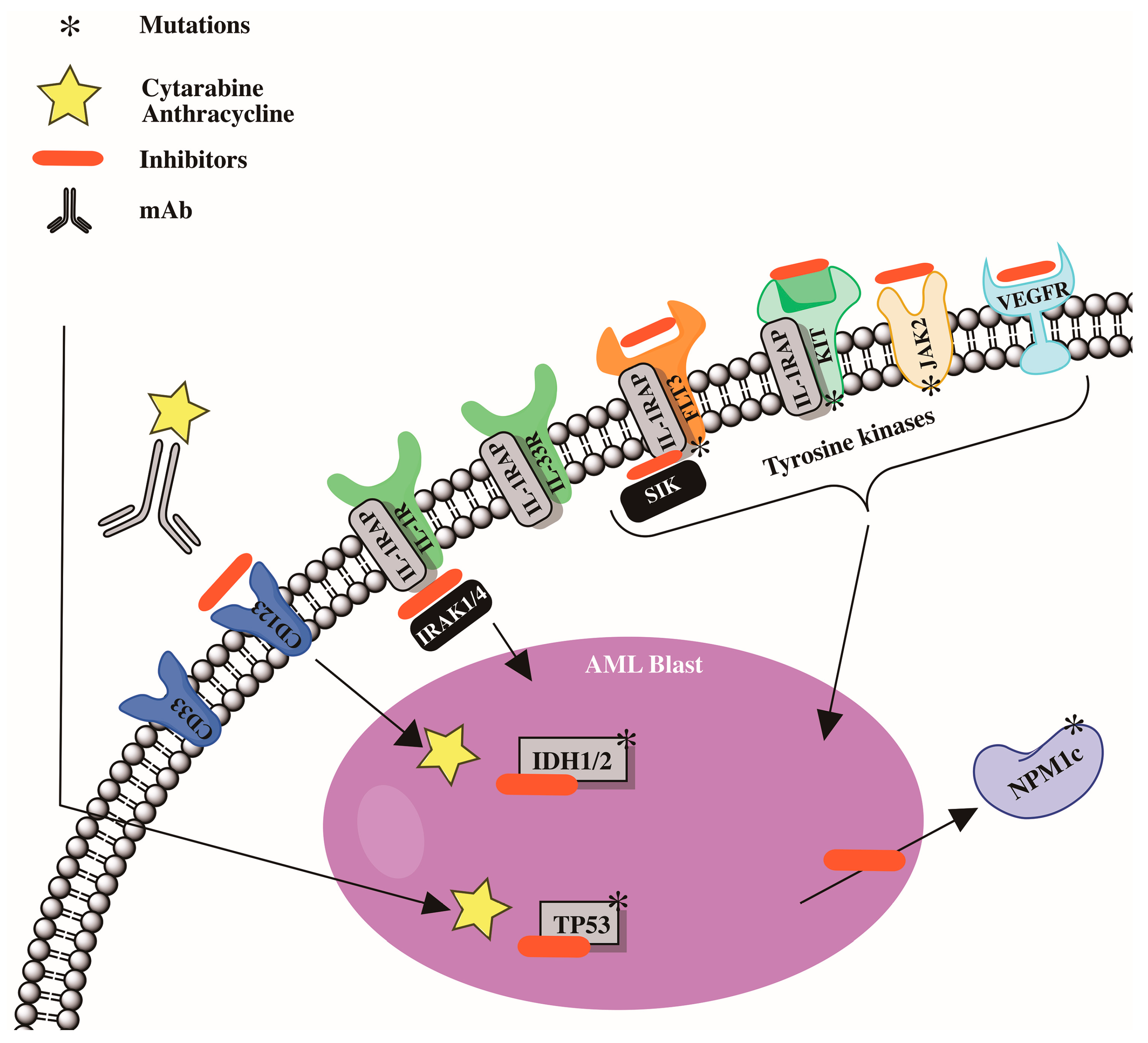
Figure 1. Cytogenetic abnormalities and mutations involved in development in acute myeloid leukemia (AML).
References
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441.
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Zeidan, A.M. Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges. Blood Rev. 2019, 36, 70–87.
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; IARC Press: Lyon, France, 2017; pp. 110–144.
- Jaffe, E.S.; Harris, N.L.; Stein, H.; Vardiman, J.W. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues, 3rd ed.; IARC Press: Lyon, France, 2001; pp. 75–105.
- DiNardo, C.D.; Cortes, J.E. Mutations in AML: Prognostic and therapeutic implications. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 348–355.
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221.
- Badar, T.; Patel, K.P.; Thompson, P.A.; DiNardo, C.; Takahashi, K.; Cabrero, M.; Borthakur, G.; Cortes, J.; Konopleva, M.; Kadia, T.; et al. Detectable FLT3-ITD or RAS mutation at the time of transformation from MDS to AML predicts for very poor outcomes. Leuk. Res. 2015, 39, 1367–1374.
- Burgess, M.R.; Hwang, E.; Firestone, A.J.; Huang, T.; Xu, J.; Zuber, J.; Bohin, N.; Wen, T.; Kogan, S.C.; Haigis, K.M.; et al. Preclinical efficacy of MEK inhibition in Nras-mutant AML. Blood 2014, 124, 3947–3955.
- Borthakur, G.; Popplewell, L.; Boyiadzis, M.; Foran, J.; Platzbecker, U.; Vey, N.; Walter, R.B.; Olin, R.; Raza, A.; Giagounidis, A.; et al. Activity of the oral mitogen-activated protein kinase kinase inhibitor trametinib in RASmutant relapsed or refractory myeloid malignancies. Cancer 2016, 122, 1871–1879.
- Pollard, J.A.; Alonzo, T.A.; Gerbing, R.B.; Ho, P.A.; Zeng, R.; Ravindranath, Y.; Dahl, G.; Lacayo, N.J.; Becton, D.; Chang, M.; et al. Prevalence and prognostic significance of KIT mutations in pediatric patients with core binding factor AML enrolled on serial pediatric cooperative trials for de novo AML. Blood 2010, 115, 2372–2379.
- Klein, K.; Kaspers, G.; Harrison, C.J.; Beverloo, H.B.; Reedijk, A.; Bongers, M.; Cloos, J.; Pession, A.; Reinhardt, D.; Zimmerman, M.; et al. Clinical impact of additional cytogenetic aberrations, cKIT and RAS mutations, and treatment elements in pediatric t(8;21)-AML: Results from an international retrospective study by the International Berlin-Frankfurt-Munster Study Group. J. Clin. Oncol. 2015, 33, 4247–4258.
- Marcucci, G.; Geyer, S.; Zhao, W.; Caroll, A.J.; Bucci, D.; Uy, G.L.; Blum, W.; Pardee, T.; Wetzler, M.; Stock, W.; et al. Adding KIT inhibitor dasatinib (DAS) to chemotherapy overcomes the negative impact of KIT mutation/over-expression in core binding factor (CBF) acute myeloid leukemia (AML): Results from CALGB 10801 (Alliance). Blood 2014, 124, 8.
- Vakiti, A.; Mewawalla, P. Acute Myeloid Leukemia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022.
- Sun, L.; Babushok, D.V. Secondary myelodysplastic syndrome and leukemia in acquired aplastic anemia and paroxysmal nocturnal hemoglobinuria. Blood 2020, 136, 36–49.
- Hasle, H. Myelodysplastic and myeloproliferative disorders of childhood. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 598–604.
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498.
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487.
- Döhner, K.; Schlenk, R.F.; Habdank, M.; Scholl, C.; Rücker, F.G.; Corbacioglu, A.; Bullinger, L.; Fröhling, S.; Döhner, H. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: Interaction with other gene mutations. Blood 2005, 106, 3740–3746.
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 2005, 352, 254–266.
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405.
- Wouters, B.J.; Löwenberg, B.; Erpelinck-Verschueren, C.A.; van Putten, W.L.; Valk, P.J.; Delwel, R. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood 2009, 113, 3088–3091.
- Pabst, T.; Eyholzer, M.; Fos, J.; Mueller, B.U. Heterogeneity within AML with CEBPA mutations; only CEBPA double mutations, but not single CEBPA mutations are associated with favourable prognosis. Br. J. Cancer 2009, 100, 1343–1346.
- Gaidzik, V.I.; Teleanu, V.; Papaemmanuil, E.; Weber, D.; Paschka, P.; Hahn, J.; Wallrabenstein, T.; Kolbinger, B.; Köhne, C.H.; Horst, H.A.; et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia 2016, 30, 2160–2168.
- Jacob, B.; Osato, M.; Yamashita, N.; Wang, C.Q.; Taniuchi, I.; Littman, D.R.; Asou, N.; Ito, Y. Stem cell exhaustion dueto Runx1 deficiency is prevented by Evi5 activation in leukemogenesis. Blood 2010, 115, 1610–1620.
- Mendler, J.H.; Maharry, K.; Radmacher, M.D.; Mrózek, K.; Becker, H.; Metzeler, K.H.; Schwind, S.; Whitman, S.P.; Khalife, J.; Kohlschmidt, J.; et al. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and microRNA expression signatures. J. Clin. Oncol. 2012, 30, 3109–3118.
- Wlodarski, M.W.; Hirabayashi, S.; Pastor, V.; Starý, J.; Hasle, H.; Masetti, R.; Dworzak, M.; Schmugge, M.; van den Heuvel-Eibrink, M.; Ussowicz, M.; et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood 2016, 127, 1387–1397.
- Green, C.L.; Tawana, K.; Hills, R.K.; Bödör, C.; Fitzgibbon, J.; Inglott, S.; Ancliff, P.; Burnett, A.K.; Linch, D.C.; Gale, R.E. GATA2 mutations in sporadic and familial acute myeloid leukaemia patients with CEBPA mutations. Br. J. Haematol. 2013, 161, 701–705.
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376.
- Devillier, R.; Mansat-De Mas, V.; Gelsi-Boyer, V.; Demur, C.; Murati, A.; Corre, J.; Prebet, T.; Bertoli, S.; Brecqueville, M.; Arnoulet, C.; et al. Role of ASXL1and TP53 mutations in the molecular classification and prognosis of acute myeloid leukemias with myelodysplasia-related changes. Oncotarget 2015, 6, 8388–8396.
- Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012, 119, 2114–2121.
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 2016, 16, 413–430.
- Lee, S.C.; Dvinge, H.; Kim, E.; Cho, H.; Micol, J.B.; Chung, Y.R.; Durham, B.H.; Yoshimi, A.; Kim, Y.J.; Thomas, M.; et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat. Med. 2016, 22, 672–678.
- Han, C.; Gao, X.; Li, Y.; Zhang, J.; Yang, E.; Zhang, L.; Yu, L. Characteristics of Cohesin Mutation in Acute Myeloid Leukemia and Its Clinical Significance. Front. Oncol. 2021, 11, 579881.
- Zhang, N.; Jiang, Y.; Mao, Q.; Demeler, B.; Tao, Y.J.; Pati, D. Characterization of the interaction between the cohesin subunits Rad21 and SA1/2. PLoS ONE 2013, 8, e69458.
- Berenstein, R. Class III receptor tyrosine kinases in acute leukemia—Biological functions and modern laboratory analysis. Biomark. Insights 2015, 10, BMI-S22433.
- Brandão, L.; Migdall-Wilson, J.; Eisenman, K.; Graham, D.K. TAM receptors in leukemia: Expression, signaling, and therapeutic implications. Crit. Rev. Oncog. 2011, 16, 47–63.
- Lee-Sherick, A.B.; Eisenman, K.M.; Sather, S.; McGranahan, A.; Armistead, P.M.; McGary, C.S.; Hunsucker, S.A.; Schlegel, J.; Martinson, H.; Cannon, C.; et al. Aberrant Mer receptor tyrosine kinase expression contributes to leukemogenesis in acute myeloid leukemia. Oncogene 2013, 32, 5359–5368.
- Rochlitz, C.; Lohri, A.; Bacchi, M.; Schmidt, M.; Nagel, S.; Fopp, M.; Fey, M.F.; Herrmann, R.; Neubauer, A. Axl expression is associated with adverse prognosis and with expression of Bcl-2 and CD34 in de novo acute myeloid leukemia (AML): Results from a multicenter trial of the Swiss Group for Clinical Cancer Research (SAKK). Leukemia 1999, 13, 1352–1358.
- Ben-Batalla, I.; Schultze, A.; Wroblewski, M.; Erdmann, R.; Heuser, M.; Waizenegger, J.S.; Riecken, K.; Binder, M.; Schewe, D.; Sawall, S.; et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine crosstalk of leukemia cells with bone marrow stroma. Blood 2013, 122, 2443–2452.
- Crosier, P.S.; Hall, L.R.; Vitas, M.R.; Lewis, P.M.; Crosier, K.E. Identification of a novel receptor tyrosine kinase expressed in acute myeloid leukemic blasts. Leuk. Lymphoma 1995, 18, 443–449.
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074.
- Li, S.; Garrett-Bakelman, F.E.; Chung, S.S.; Sanders, M.A.; Hricik, T.; Rapaport, F.; Patel, J.; Dillon, R.; Vijay, P.; Brown, A.L.; et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat. Med. 2016, 22, 792–799.
- Li, S.; Mason, C.; Melnick, A. Genetic and epigenetic heterogeneity in acute myeloid leukemia. Curr. Opin. Genet. Dev. 2016, 36, 100–106.
- Guillamot, M.; Cimmino, L.; Aifantis, I. The impact of DNA methylation in hematopoietic malignancies. Trends Cancer 2016, 2, 70–83.
- Hassan, C.; Afshinnekoo, E.; Li, S.; Wu, S.; Mason, C.E. Genetic and epigenetic heterogeneity and the impact on cancer relapse. Exp. Hematol. 2017, 54, 26–30.
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398.
- Koh, H.J.; Lee, S.M.; Son, B.G.; Lee, S.H.; Ryoo, Z.Y.; Chang, K.T.; Park, J.W.; Park, D.C.; Song, B.J.; Veech, R.L.; et al. Cytosolic NADP+-dependent isocitrate dehydrogenase plays a key role in lipid metabolism. J. Biol. Chem. 2004, 279, 39968–39974.
- Joseph, J.W.; Jensen, M.V.; Ilkayeva, O.; Palmieri, F.; Alárcon, C.; Rhodes, C.J.; Newgard, C.B. The mitochondrial citrate/isocitrate carrier plays a regulatory role in glucose-stimulated insulin secretion. J. Biol. Chem. 2006, 281, 35624–35632.
- Cairns, R.A.; Mak, T.W. Oncogenic isocitrate dehydrogenase mutations: Mechanisms, models, and clinical opportunities. Cancer Discov. 2013, 3, 730–741.
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744.
- Losman, J.A.; Looper, R.E.; Koivunen, P.; Lee, S.; Schneider, R.K.; McMahon, C.; Cowley, G.S.; Root, D.E.; Ebert, B.L.; Kaelin, W.G., Jr. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013, 339, 1621–1625.
- Becker, H.; Marcucci, G.; Maharry, K.; Radmacher, M.D.; Mrózek, K.; Margeson, D.; Whitman, S.P.; Wu, Y.Z.; Schwind, S.; Paschka, P.; et al. Favorable prognostic impact of NPM1 mutations in older patients with cytogenetically normal de novo acute myeloid leukemia and associated gene- and microRNA-expression signatures: A Cancer and Leukemia Group B study. J. Clin. Oncol. 2010, 28, 596–604.
- Alpermann, T.; Schnittger, S.; Eder, C.; Dicker, F.; Meggendorfer, M.; Kern, W.; Schmid, C.; Aul, C.; Staib, P.; Wendtner, C.M.; et al. Molecular subtypes of NPM1 mutations have different clinical profiles, specific patterns of accompanying molecular mutations and varying outcomes in intermediate risk acute myeloid leukemia. Haematologica 2016, 101, e55–e58.
- Jalagadugula, G.; Mao, G.; Kaur, G.; Goldfinger, L.E.; Dhanasekaran, D.N.; Rao, A.K. Regulation of platelet myosin light chain (MYL9) by RUNX1: Implications for thrombocytopenia and platelet dysfunction in RUNX1 haplodeficiency. Blood 2010, 116, 6037–6045.
- Jalagadugula, G.; Mao, G.; Kaur, G.; Dhanasekaran, D.N.; Rao, A.K. Platelet protein kinase C-theta deficiency with human RUNX1 mutation: PRKCQ is a transcriptional target of RUNX1. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 921–927.
- Kaur, G.; Jalagadugula, G.; Mao, G.; Rao, A.K. RUNX1/core binding factor A2 regulates platelet 12-lipoxygenase gene (ALOX12): Studies in human RUNX1 haplodeficiency. Blood 2010, 115, 3128–3135.
- Antony-Debre, I.; Bluteau, D.; Itzykson, R.; Baccini, V.; Renneville, A.; Boehlen, F.; Morabito, M.; Droin, N.; Deswarte, C.; Chang, Y.; et al. MYH10 protein expression in platelets as a biomarker of RUNX1 and FLI1 alterations. Blood 2012, 120, 2719–2722.
- Bluteau, D.; Balduini, A.; Balayn, N.; Currao, M.; Nurden, P.; Deswarte, C.; Leverger, G.; Noris, P.; Perrotta, S.; Solary, E.; et al. Thrombocytopenia-associated mutations in the ANKRD26 regulatory region induce MAPK hyperactivation. J. Clin. Investig. 2014, 124, 580–591.
- Paz-Priel, I.; Friedman, A. C/EBPalpha dysregulation in AML and ALL. Crit. Rev. Oncog. 2011, 16, 93–102.
- Tsai, F.Y.; Keller, G.; Kuo, F.C.; Weiss, M.; Chen, J.; Rosenblatt, M.; Alt, F.W.; Orkin, S.H. An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature 1994, 371, 221–226.
- Rodrigues, N.P.; Janzen, V.; Forkert, R.; Dombkowski, D.M.; Boyd, A.S.; Orkin, S.H.; Enver, T.; Vyas, P.; Scadden, D.T. Haploinsufficiency of GATA-2 perturbs adult hematopoietic stem-cell homeostasis. Blood 2005, 106, 477–484.
- Kazenwadel, J.; Secker, G.A.; Liu, Y.J.; Rosenfeld, J.A.; Wildin, R.S.; Cuellar-Rodriguez, J.; Hsu, A.P.; Dyack, S.; Fernandez, C.V.; Chong, C.E.; et al. Loss-of-function germline GATA2 mutations in patients with MDS/AML or MonoMAC syndrome and primary lymphedema reveal a key role for GATA2 in the lymphatic vasculature. Blood 2012, 119, 1283–1291.
- Godley, L.A. Inherited predisposition to acute myeloid leukemia. Semin. Hematol. 2014, 51, 306–321.
- Solomon, D.A.; Kim, J.S.; Waldman, T. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science 2011, 333, 1039–1043.
- Losada, A. Cohesin in cancer: Chromosome segregation and beyond. Nat. Rev. Cancer 2014, 14, 389–393.
- Litwin, I.; Wysocki, R. New insights into cohesin loading. Curr. Genet. 2018, 64, 53–61.
- Dauban, L.; Montagne, R.; Thierry, A.; Lazar-Stefanita, L.; Bastié, N.; Gadal, O.; Cournac, A.; Koszul, R.; Beckouët, F. Regulation of Cohesin-Mediated Chromosome Folding by Eco1 and Other Partners. Mol. Cell 2020, 77, 1279–1293.e4.
- Cuartero, S.; Innes, A.J.; Merkenschlager, M. Towards a Better Understanding of Cohesin Mutations in AML. Front. Oncol. 2019, 9, 867.
- Kagey, M.H.; Newman, J.J.; Bilodeau, S.; Zhan, Y.; Orlando, D.; Am van Berkumm, N.L.; Ebmeier, C.C.; Goossens, J.; Rahl, P.B.; Levine, S.S.; et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010, 467, 430–435.
- Guillou, E.; Ibarra, A.; Coulon, V.; Casado-Vela, J.; Rico, D.; Casal, I.; Schwob, E.; Losada, A.; Méndez, J. Cohesin organizes chromatin loops at DNA replication factories. Genes Dev. 2010, 24, 2812–2822.
- McAleenan, A.; Clemente-Blanco, A.; Cordon-Preciado, V.; Sen, N.; Esteras, M.; Jarmuz, A.; Aragón, L. Post-replicative repair involves separase-dependent removal of the kleisin subunit of cohesin. Nature 2012, 493, 250–254.
- Panigrahi, A.K.; Pati, D. Higher-order orchestration of hematopoiesis: Is cohesin a new player? Exp. Hematol. 2012, 40, 967–973.
- Guo, Y.; Monahan, K.; Wu, H.; Gertz, J.; Varley, K.E.; Li, W.; Myers, R.M.; Maniatis, T.; Wu, Q. CTCF/cohesin-mediated DNA looping is required for protocadherin alpha promoter choice. Proc. Natl. Acad. Sci. USA 2012, 109, 21081–21086.
- Haarhuis, J.H.I.; van der Weide, R.H.; Blomen, V.A.; Yáñez-Cuna, J.O.; Amendola, M.; van Ruiten, M.S.; Krijger, P.H.L.; Teunissen, H.; Medema, R.H.; van Steensel, B.; et al. The cohesin release factor wapl restricts chromatin loop extension. Cell 2017, 169, 693–707.
- Schwarzerm, W.; Abdennur, N.; Goloborodko, A.; Pekowska, A.; Fudenberg, G.; Loe-Mie, Y.; Fonseca, N.A.; Huber, W.; Haering, C.H.; Mirny, L.; et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature 2017, 551, 51–56.
- Rao, S.S.; Huang, S.C.; Glenn St Hilaire, B.; Engreitz, J.M.; Perez, E.M.; Kieffer-Kwon, K.R.; Sanborn, A.L.; Johnstone, S.E.; Bascom, G.D.; Bochkov, I.D.; et al. Cohesin loss eliminates all loop domains. Cell 2017, 171, 305–320.
- Bintu, B.; Mateo, L.J.; Su, J.H.; Sinnott-Armstrong, N.A.; Parker, M.; Kinrot, S.; Yamaya, K.; Boettiger, A.N.; Zhuang, X. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 2018, 362, eaau1783.
- National Comprehensive Cancer Network. NCCN Guidelines & Clinical Resources. Adapted from the National Cancer Centers Network (NCCN). 2020. Available online: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1410 (accessed on 14 December 2022).
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719.
- Grimwade, D.; Hills, R.K.; Moorman, A.V.; Walker, H.; Chatters, S.; Goldstone, A.H.; Wheatley, K.; Harrison, C.J.; Burnett, A.K.; National Cancer Research Institute Adult Leukaemia Working Group. Refinement of cytogenetic classification in acute myeloid leukemia: Determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010, 116, 354–365.
- Schoch, C.; Schnittger, S.; Klaus, M.; Kern, W.; Hiddemann, W.; Haferlach, T. AML with 11q23/MLL abnormalities as defined by the WHO classification: Incidence, partner chromosomes, FAB subtype, age distribution, and prognostic impact in an unselected series of 1897 cytogenetically analyzed AML cases. Blood 2003, 102, 2395–2402.
- Ayatollahi, H.; Bazi, A.; Sadeghian, M.H.; Fani, A.; Siyadat, P.; Sheikhi, M.; Sargazi-aval, O. The Survival of Patients with t(15;17)(q22;q12) Positive Acute Promyelocytic Leukemia: A Study in North-East of Iran. Iran. J. Pathol. 2020, 15, 175–181.
- Eghtedar, A.; Borthakur, G.; Ravandi, F.; Jabbour, E.; Cortes, J.; Pierce, S.; Kantarjian, H.; Garcia-Manero, G. Characteristics of translocation (16;16)(p13;q22) acute myeloid leukemia. Am. J. Hematol. 2012, 87, 317.
- Rücker, F.G.; Agrawal, M.; Corbacioglu, A.; Weber, D.; Kapp-Schwoerer, S.; Gaidzik, V.I.; Jahn, N.; Schroeder, T.; Wattad, M.; Lübbert, M.; et al. Measurable residual disease monitoring in acute myeloid leukemia with t(8;21)(q22;q22.1): Results from the AML Study Group. Blood 2019, 134, 1608–1618.
- Stölzel, F.; Mohr, B.; Kramer, M.; Oelschlägel, U.; Bochtler, T.; Berdel, W.E.; Kaufmann, M.; Baldus, C.D.; Schäfer-Eckart, K.; Stuhlmann, R.; et al. Karyotype complexity and prognosis in acute myeloid leukemia. Blood Cancer J. 2016, 6, e386.
- Groschel, S.; Schlenk, R.F.; Engelmann, J.; Rockova, V.; Teleanu, V.; Kühn, M.W.; Eiwen, K.; Erpelinck, C.; Havermans, M.; Lübbert, M.; et al. Deregulated expression of EVI1 defines a poor prognostic subset of MLL-rearranged acute myeloid leukemias: A study of the German-Austrian Acute Myeloid Leukemia Study Group and the Dutch-Belgian-Swiss HOVON/SAKK Cooperative Group. J. Clin. Oncol. 2013, 31, 95–103.
- Liu, K.; Tirado, C.A. MECOM: A Very Interesting Gene Involved also in Lymphoid Malignancies. J. Assoc. Genet. Technol. 2019, 45, 109–114.
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017b. ELN recommendations from an international expert panel. Blood 2017, 129, 424–447.
- Yu, J.; Li, Y.; Li, T.; Li, Y.; Xing, H.; Sun, H.; Sun, L.; Wan, D.; Liu, Y.; Xie, X.; et al. Gene mutational analysis by NGS and its clinical significance in patients with myelodysplastic syndrome and acute myeloid leukemia. Exp. Hematol. Oncol. 2020, 6, 2.
- Han, X.; Li, W.; He, N.; Feng, P.; Pang, Y.; Ji, C.; Ma, D. Gene mutation patterns of Chinese acute myeloid leukemia patients by targeted next-generation sequencing and bioinformatic analysis. Clin. Chim. Acta 2018, 479, 25–37.
- Chung, W.; Kelly, A.D.; Kropf, P.; Fung, H.; Jelinek, J.; Su, X.Y.; Roboz, G.J.; Kantarjian, H.M.; Azab, M.; Issa, J.P.J. Genomic and epigenomic predictors of response to guadecitabine in relapsed/refractory acute myelogenous leukemia. Clin. Epigenet. 2019, 11, 106.
- Yang, L.; Shen, K.; Zhang, M.; Zhang, W.; Cai, H.; Lin, L.; Long, X.; Xing, S.; Tang, Y.; Xiong, J.; et al. Clinical features and microRNA expression patterns between AML patients with DNMT3A R882 and frameshift mutations. Front Oncol. 2019, 24, 1133.
- Folta, A.; Culen, M.; Jeziskova, I.; Herudkova, Z.; Tom, N.; Hlubinkova, T.; Janeckova, V.; Durinikova, A.; Vydra, J.; Semerad, L.; et al. Prognostic significance of mutation profile at diagnosis and mutation persistence during disease remission in adult acute myeloid leukaemia patients. Br. J. Haematol. 2019, 186, 300–310.
- Vetro, C.; Haferlach, T.; Meggendorfer, M.; Stengel, A.; Jeromin, S.; Kern, W.; Haferlach, C. Cytogenetic and molecular genetic characterization of KMT2A-PTD positive acute myeloid leukemia in comparison to KMT2A-rearranged acute myeloid leukemia. Cancer Genet. 2020, 240, 15–22.
- Antar, A.I.; Otrock, Z.K.; Jabbour, E.; Mohty, M.; Bazarbachi, A. FLT3 inhibitors in acute myeloid leukemia: Ten frequently asked questions. Leukemia 2020, 34, 682–696.
- Darracq, A.; Pak, H.; Bourgoin, V.; Zmiri, F.; Dellaire, G.; Affar, E.B.; Milot, E. NPM and NPM-MLF1 interact with chromatin remodeling complexes and influence their recruitment to specific genes. PLoS Genet. 2019, 15, e1008463.
- Patel, S.S.; Kuo, F.C.; Gibson, C.J.; Steensma, D.P.; Soiffer, R.J.; Alyea, E.P., 3rd; Chen, Y.A.; Fathi, A.T.; Graubert, T.A.; Brunner, A.M.; et al. High NPM1-mutant allele burden at diagnosis predicts unfavorable outcomes in de novo AML. Blood 2018, 131, 2816–2825.
- Cucchi, D.G.J.; Denys, B.; Kaspers, G.J.L.; Janssen, J.J.W.M.; Ossenkoppele, G.J.; de Haas, V.; Zwaan, C.M.; van den Heuvel-Eibrink, M.M.; Philippé, J.; Csikós, T.; et al. RNA-based FLT3-ITD allelic ratio is associated with outcome and ex vivo response to FLT3 inhibitors in pediatric AML. Blood 2018, 131, 2485–2489.
- Boileau, M.; Shirinian, M.; Gayden, T.; Harutyunyan, A.S.; Chen, C.C.L.; Mikael, L.G.; Duncan, H.M.; Neumann, A.L.; Arreba-Tutusaus, P.; De Jay, N.; et al. Mutant H3 histones drive human pre-leukemic hematopoietic stem cell expansion and promote leukemic aggressiveness. Nat. Commun. 2019, 10, 2891.
- Tallman, M. Prognostic significance of molecular markers and targeted regimens in the management of acute myeloid leukemia. J. Natl. Compr. Cancer Netw. 2018, 16, 656–659.
- Tallman, M.S.; Wang, E.S.; Altman, J.K.; Appelbaum, F.R.; Bhatt, V.R.; Bixby, D.; Coutre, S.E.; De Lima, M.; Fathi, A.T.; Fiorella, M.; et al. Acute myeloid leukemia, version 3.2019, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. 2019, 17, 721–749.
- Corces-Zimmerman, M.R.; Hong, W.J.; Weissman, I.L.; Medeiros, B.C.; Majeti, R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc. Natl. Acad. Sci. USA 2014, 111, 2548–2553.
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333.
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478.
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Bourgoin, V.; Mollica, L.; Dubé, M.P.; Busque, L. Lineage restriction analyses in CHIP indicate myeloid bias for TET2 and multipotent stem cell origin for DNMT3A. Blood 2018, 132, 277–280.
More
Information
Subjects:
Cell Biology; Pathology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.8K
Revisions:
2 times
(View History)
Update Date:
30 Mar 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No