Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Cristian Sandoval and Version 2 by Camila Xu.
Acute myeloid leukemia is a cancerous condition that affects hemopoietic stem cells or progenitors and is defined by the stopping of myeloid lineage development and abnormal proliferation.
- adults
- cytochrome
- monooxygenase
- polymorphisms
1. Introduction
Acute myeloid leukemia is a cancerous condition that affects hemopoietic stem cells or progenitors and is defined by the stopping of myeloid lineage development and abnormal proliferation [1]. The most prevalent acute leukemia in adults is acute myeloid leukemia, which has a wide range of genetic variations. In 2021, more than 20,000 new cases of acute myeloid leukemia were expected in the US [2]. Traditionally, acute myeloid leukemia has been categorized based on immunophenotype and morphology. However, genetic abnormalities, such as chromosomal translocations and transcription factor involvement, must be considered in acute myeloid leukemia diagnostic algorithms [3][4][3,4]. These factors led to the classification of acute myeloid leukemia into six groups [3]: myeloid proliferations linked to Down syndrome, myeloid sarcoma, recurring genetic abnormalities, therapy-related myeloid neoplasms, and acute myeloid leukemia with myelodysplasia-related alterations.
2. Pathophysiology of Acute Myeloid Leukemia
2.1. Cytogenetic Abnormalities
Acute myeloid leukemia is characterized by mutations in hematopoiesis-related genes [5][24]. Ineffective erythropoiesis and bone marrow failure are caused by these mutations, which cause a clonal increase in undifferentiated myeloid progenitors (blasts) in the peripheral blood and bone marrow. Recent research has suggested that it could result from several recurring genetic changes in hematopoietic stem cells that accumulate over time [6][7][8][9][10][11][12][15,25,26,27,28,29,30]. Acute myeloid leukemia often develops from scratch in a previously healthy person. Although the precise source of genetic abnormalities is unknown, a few known risk factors include smoking, chemotherapy, and radiation exposure [13][31]. Aplastic anemia, paroxysmal nocturnal hemoglobinuria, myelodysplastic syndrome, and myeloproliferative diseases can all develop into acute myeloid leukemia [14][15][32,33].
Genetic mutations that have familial causes should also be considered (Table 1). The most prevalent mutational subset in acute myeloid leukemia is type 1 mutations, which are present in about two-thirds of patients and result in abnormal activation and proliferation of cellular signaling pathways (e.g., FMS-like tyrosine kinase 3 (FLT3); Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS); NRAS Proto-Oncogene, GTPase (NRAS); Tyrosine-protein phosphatase non-receptor type 11 (PTPN11); neurofibromin 1 (NF1); and KIT proto-oncogene, receptor tyrosine kinase (KIT)). It is interesting to note that mutations in this class are usually found in subclonal cellular fractions, indicating that they are frequently late clonal events in the development of illness [6][15].
Table 1.
Recurrent mutations in acute myeloid leukemia.
| Functional Class | Specific Example Mutations | References |
|---|---|---|
| Signaling and kinase pathways | FLT3, KRAS, NRAS, KIT, PTPN11, and NF1 | [6][15] |
| DNA methylation and chromatin modification | DNMT3A, IDH1, IDH2, TET2, ASXL1, EZH2, and MLL/KMT2A | [6][16][17][15,34,35] |
| Nucleophosmin | NPM1 | [18][19][36,37] |
| Transcription factors | CEBPA, RUNX1, and GATA2 | [20][21][22][23][24]][38,39[,4025],41[26],42[,4327,44,45] |
| Tumor suppressors | TP53 | [28][29][30][46,47,48] |
| Spliceosome complex | SRSF2, U2AF1, SF3B1, and ZRSR2 | [31][32][49,50] |
| Cohesin complex | RAD21, STAG1, STAG2, SMC1A, and SMC3 | [33][34][51,52] |
Somatic mutations within key epigenetic regulators are identified in >50% of acute myeloid leukemia, and are now recognized as a key, and often an inciting, component of leukemogenesis [6][15]. It is of interest that age-related clonal hematopoiesis, identified in >10% of individuals over age 65, is predominantly defined by the clonal outgrowth of preleukemic clones harboring mutations in one of the genes within this epigenetic class [16][17][34,35].
Along with FLT3 and DNA methyltransferase 3 alpha (DNMT3A; https://www.ncbi.nlm.nih.gov/gene/1788; accessed on 14 January 2023), nucleophosmin 1 (NPM1) is one of the three most frequent driver mutations in acute myeloid leukemia. The regulation of pathways for cell proliferation, differentiation, adhesion, and death by receptor tyrosine kinase (RTKs) signaling pathways in acute myeloid leukemia is crucial for the onset and spread of malignancy. Around 20 separate subfamilies make up the RTKs, which include class III and TYRO3, AXL, and MERTK (TAM) family RTKs [35][53]. Class III RTKs, which include c-Kit, CSF1R, FLT3, and platelet-derived growth factor receptors (PDGFR), have been discovered to have a major effect on leukemogenesis and transformation into acute myeloid leukemia. Class III RTKs have been linked to aberrant activation that promotes proliferation in leukemia. Particularly, FLT3 expression and c-Kit mutations are critical for acute myeloid leukemia. Both RTKs have been crucial targets in the development of antileukemic therapies, since they are both associated with worse prognoses. TYRO3, AXL, and MERTK are members of the TAM family of RTKs. These are essential for platelet activation and stabilization, for the normal hematopoiesis of certain innate immune cells, and have been linked to erythropoiesis [19][37]. TAM RTKs are critical for normal hematopoietic development, but they can activate pathways for proliferation and survival in cancer cells, particularly in acute leukemia [36][54]. TAM RTKs, particularly AXL and MERTK, have been linked to hematologic malignancies and have grown in interest as potential targets for creating novel treatments [36][37][38][39][40][54,55,56,57,58].
In addition, compared to the genomes of other cancers, acute myeloid leukemia genomes are typically less mutated [41][59], with similar distributions of mutations before and following relapse [42][43][60,61]. Of these mutations, many frequently occur in genes involved in DNA methylation and epigenetic regulation, such as DNMT3A, TET1/2, and IDH1/2 [44][62]. Hassan et al. [45][63] point to a greater need for understanding acute myeloid leukemia through a non-genetic lens, focusing on DNA methylation and other epigenetic modalities. They also suggest relative independence between the progression of acute myeloid leukemia and the disease’s strictly genetic landscape.
IDH1 gene mutations are present in around 6–10% of individuals with acute myeloid leukemia [46][64]. Given its capacity to create cytoplasmic NADPH and glucose sensing, IDH1 is implicated in controlling cellular metabolism, particularly lipid metabolism [47][48][65,66]. Isocitrate is oxidized to α-ketoglutarate by wild-type isocitrate dehydrogenases [49][67]. It has been hypothesized that the IDH1 Arg132 mutation alters the way the enzyme functions, causing α-ketoglutarate to be converted to R(—)-2-hydroxyglutarate [50][68]. This excess of R(—)-2-hydroxyglutarate causes cellular proliferation to rise and cellular differentiation to be compromised [51][69].
Nucleophosmin 1 mutations, occurring almost exclusively within exon 12 of the gene, occur in approximately one-third of adults with acute myeloid leukemia, and in more than 50% of NK-AML. The NPM1 gene encodes for the nuclear chaperone protein NPM, which shuttles between the nucleus and cytoplasm and plays a role in diverse cellular functions, including protein formation, ribosome biogenesis, DNA replication, and the cell cycle. NPM1 mutations are typically stable throughout the disease course, are identified in nearly all leukemic cells, and impart a distinct expression profile [52][70]. NPM1 mutations in the setting of mutant DNMT3A, particularly in the setting of FLT3 internal tandem duplication (FLT3-ITD), confer a markedly poor prognosis [6][53][15,71].
Approximately 20% to 25% of adults with acute myeloid leukemia have mutations involving the myeloid transcription factors Runt-related transcription factor 1 (RUNX1), CEBPA, and/or GATA binding protein 2 (GATA2) [5][24]. In this sense, RUNX1 is known to act as a direct transcriptional activator of several proteins important for platelet function and as a transcriptional repressor of others, including MYH10 [54][55][56][57][72,73,74,75] and ANKRD26 [58][76]. In addition, CEBPA encodes a master hematopoietic transcription factor that acts as a critical regulator of granulocyte and monocyte differentiation [59][77], while GATA2 encodes a zinc finger transcription factor critical for normal hematopoiesis [60][61][78,79] and lymphatic vascular development [62][63][80,81].
Tumor protein p53 (TP53) is a key tumor suppressor with highly variable functions related to the maintenance of genomic stability, including regulation of cellular senescence, apoptosis, metabolism, and DNA repair. Although uncommon in de novo acute myeloid leukemia, TP53 mutations occur in ~15% of therapy-related acute myeloid leukemia or acute myeloid leukemia with myelodysplastic syndrome-related changes, and are predominantly associated with complex cytogenetics, advanced age, chemotherapy resistance, and poor survival [29][30][47,48]. Irrespective of age or treatment modality, TP53 mutations in acute myeloid leukemia portend lower response rates and inferior outcomes compared with TP53 wild-type acute myeloid leukemia patients [31][49].
Frequently mutated in myelodysplastic syndrome and myeloproliferative neoplasms, mutations in splicing factors (SF3B1, SRSF2, U2AF1, and ZRSR2) are identified in ~10% of patients with acute myeloid leukemia and are associated with older age, less proliferative disease, poor rates of response to standard treatment, and decreased survival. Spliceosome mutations are postulated to promote malignancy through the missplicing of various genes involved in epigenetic regulation, transcription, and genome integrity [32][50].
The structural maintenance of chromosomes (SMC3 and SMC1A), RAD cohesin complex component (RAD21), and cohesin subunit SA (STAG1/STAG2) make up the four core elements of cohesin with a ring shape. Cohesin helps several other subunits, such as NIPBL, MAU2, WAPL, PDS5A, PDS5B, and sororin, to form cohesion during the cell cycle [64][65][66][67][82,83,84,85]. Consequently, the ring-shaped cohesin controls the sister chromatids’ separation, DNA replication, and repair of the broken double-strand DNA during the advancement of the cell cycle [68][69][70][71][72][86,87,88,89,90]. To control chromatin structure and gene expression, the cohesin complex can also interact with the transcriptional repressor CTCF, promoters, mediators, enhancers, initiation and elongation forms of RNA polymerase II (RNAPII), or transcription factors (TFs) [69][73][74][75][76][77][87,91,92,93,94,95].
Acute myeloid leukemia prognosis is exceedingly variable and unpredictable. It can be caused by molecular changes, chromosomal translocations, or genetic mutations. Genetic mutations have been shown to occur in around 97% of cases. Table 2 shows an updated classification of acute myeloid leukemia based on the National Comprehensive Cancer Network’s cytogenetic and molecular criteria [78][79][96,97]. The following are examples of cytogenetic subsets: (1) chromosomal translocations [t(15;17)(q22,q21)] and core binding factor acute myeloid leukemia (CBF-AML), both of which are cytogenetic/molecular subgroups of inversion 16 [inv16(p13;q22)] or t(16;16)(p13;q22); (2) individuals with cytogenetically normal acute myeloid leukemia (CN-AML) who have monosomy 5 or 7 or t(9 or 11) have a low risk (40–50% of patients) [67][68][69][70][71][72][85,86,87,88,89,90]; (3) individuals with t(6;9), inv (3), or 11q changes (11q23 translocations) [67][68][69][70][71][72][85,86,87,88,89,90]; (4) and those with other karyotypes have been demonstrated to have a higher risk of treatment failure and mortality [80][81][82][83][84][85][98,99,100,101,102,103]. Furthermore, people with translocations involving the MECOM (myelodysplastic syndrome-1 and ecotropic viral integration site 1 (EVI1) complex locus) gene on chromosome 3q26.2 have a very poor prognosis [86][87][104,105].
Table 2. Classification of acute myeloid leukemia based on the National Comprehensive Cancer Network’s cytogenetic and molecular criteria.
| Type | Diagnostic Criteria |
|---|---|
| Acute myeloid leukemia with minimal differentiation | Blasts are negative (<3%) for MPO and SBB. |
| Expression of two or more myeloid-associated antigens, such as CD13, CD33, and CD117. | |
| Acute myeloid leukemia without maturation | ≥3% blasts positive for MPO or SBB and negative for NSE. |
| Maturing cells of the granulocytic lineage constitute <10% of the nucleated bone marrow cells. | |
| Expression of two or more myeloid-associated antigens, such as MPO, CD13, CD33, and CD117. | |
| Acute myeloid leukemia with maturation | ≥3% blasts positive for MPO or SBB. |
| Maturing cells of the granulocytic lineage constitute ≥10% of the nucleated bone marrow cells. | |
| Monocyte lineage cells constitute <20% of bone marrow cells. | |
| Expression of two or more myeloid–associated antigens, such as MPO, CD13, CD33, and CD117. | |
| Acute basophilic leukemia | Blasts and immature/mature basophils with metachromasia on toluidine blue staining. |
| Blasts are negative for cytochemical MPO, SBB, and NSE. | |
| No expression of strong CD117 equivalent (to exclude mast cell leukemia). | |
| Acute myelomonocytic leukemia | ≥20% monocytes and their precursors. |
| ≥20% maturing granulocytic cells. | |
| ≥3% of blasts positive for MPO. | |
| Acute monocytic leukemia | ≥80% monocytes and/or their precursors (monoblasts and/or promonocytes). |
| <20% maturing granulocytic cells. | |
| Blasts and promonocytes expressing at least two monocytic markers including CD11c, CD14, CD36 and CD64, or NSE. | |
| Acute erythroid leukemia | ≥30% immature erythroid cells (proerythroblasts) |
| Bone marrow with erythroid predominance, usually ≥80% of cellularity | |
| Acute megakaryoblastic leukemia | Blasts express at least one or more of the platelet glycoproteins: CD41 (glycoprotein llb), CD61 (glycoprotein IIIa), or CD42b (glycoprotein lb) |
MPO: myeloperoxidase; NSE: nonspecific esterase–butyrate; SBB: Sudan Black B.
2.2. Mutations
Modern molecular technologies have permitted the identification of a wide spectrum of genetic disorders. Six genes have already been incorporated into the European Leukemia Net risk categories [88][5], including FLT3, NPM1, CCAAT/enhancer binding protein α (CEBPA), RUNX1, additional sexcombs-like 1 (ASXL1), and TP53 [88][5]. Other recurrent gene mutations in acute myeloid leukemia patients have been discovered [89][90][91][92][93][94][106,107,108,109,110,111]. Furthermore, research has been undertaken on the essential roles played by recurrent gene mutations in the pathogenesis of acute myeloid leukemia, as well as the development of medicines that precisely target gene alterations [95][96][97][98][99][100][101][112,113,114,115,116,117,118]. Preleukemic cell detection, particularly in acute myeloid leukemia patients with mutant DNMT3A and TET2 genes, has been linked to leukemia genesis [102][103][119,120]. Mutations in the DNMT3A and TET2 genes are common in patients with clonal hematopoiesis of undetermined potential [16][17][104][105][34,35,121,122]; these mutations may serve as markers for the identification of preleukemia cells [102][103][119,120]. A summary of the pathophysiology of acute myeloid leukemia is shown in Figure 12.
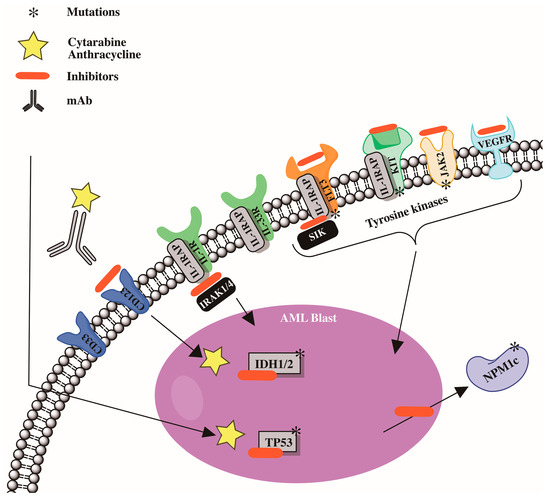
Figure 12.
Cytogenetic abnormalities and mutations involved in development in acute myeloid leukemia (AML).