Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alexander Leonardo Silva-Junior | -- | 4315 | 2023-03-28 13:58:34 | | | |
| 2 | Rita Xu | Meta information modification | 4315 | 2023-03-29 03:44:21 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Silva-Junior, A.L.; Oliveira, L.D.S.; Belezia, N.C.T.; Tarragô, A.M.; Costa, A.G.D.; Malheiro, A. Immunology in the Acute Phase of COVID-19. Encyclopedia. Available online: https://encyclopedia.pub/entry/42588 (accessed on 28 July 2026).
Silva-Junior AL, Oliveira LDS, Belezia NCT, Tarragô AM, Costa AGD, Malheiro A. Immunology in the Acute Phase of COVID-19. Encyclopedia. Available at: https://encyclopedia.pub/entry/42588. Accessed July 28, 2026.
Silva-Junior, Alexander Leonardo, Lucas Da Silva Oliveira, Nara Caroline Toledo Belezia, Andréa Monteiro Tarragô, Allyson Guimarães Da Costa, Adriana Malheiro. "Immunology in the Acute Phase of COVID-19" Encyclopedia, https://encyclopedia.pub/entry/42588 (accessed July 28, 2026).
Silva-Junior, A.L., Oliveira, L.D.S., Belezia, N.C.T., Tarragô, A.M., Costa, A.G.D., & Malheiro, A. (2023, March 28). Immunology in the Acute Phase of COVID-19. In Encyclopedia. https://encyclopedia.pub/entry/42588
Silva-Junior, Alexander Leonardo, et al. "Immunology in the Acute Phase of COVID-19." Encyclopedia. Web. 28 March, 2023.
Copy Citation
COVID-19 is a viral disease that has caused millions of deaths around the world since 2020. Many strategies have been developed to manage patients in critical conditions; comprehension of the immune system is a key factor in viral clearance, tissue repairment, and adaptive immunity stimulus. Participation of immunity has been identified as a major factor, along with biomarkers, prediction of clinical outcomes, and antibody production after infection. Immune cells have been proposed not only as a hallmark of severity, but also as a predictor of clinical outcomes, while dynamics of inflammatory molecules can also induce worse consequences for acute patients.
SARS-CoV-2
inflammation
antibodies
adaptive immunity
1. Background
Coronavirus disease 2019 (COVID-19) is an infectious disease caused by a betacoronavirus, reported as the pathogen of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). It was first described in 2019, related to transmission from wild animals, in the province of Wuhan, China. Cases rapidly increased around the world due to its easy transmission via aerosol; however, other fluids, such as urine and saliva, as well as surfaces (paper, wood, and metal) for more than 4 days [1][2][3][4], were suggested as transmission routes, but conclusive evidence is still needed.
After exposure to the virus, the respiratory tract is the main tissue affected by angiotensin-converting enzyme (ACE2) expression, the human’s receptor to the viral spike protein. After binding, SARS-CoV-2 enters the target cell, thereby establishing infection. The human first line of defense plays an important role in immune recognition and management of the disease. The viral protein receptor is expressed mainly in pulmonary, cardiac, renal, and lipid-rich tissues, and symptoms are represented by cough and fever; in severe cases, symptoms may evolve to pneumonia and death. Dynamics of extrinsic factors, such as age and comorbidities [5][6][7], together with intrinsic factors, such as immune response, have been demonstrated as key factors in a patient’s clinical outcome during the acute phase, in their post-COVID-19 symptoms, and in their protection from new variants [8][9][10].
The innate and adaptive immune responses contribute to viral clearance and to the production of specific antibodies. Many studies have evaluated and characterized the immunity imbalance in patients with active disease, showing a hyperinflammatory response in severe patients, led by a cytokine storm that causes more disadvantages than benefits in the resolution of the disease [11][12][13][14][15][16]. However, few papers have proposed evaluating those patients who had a favorable outcome, known as convalescent patients.
Many individuals may present long-term symptoms, named post-acute COVID-19 (starting 3–12 weeks from the acute phase) and post-COVID-19 syndrome (or long COVID, starting more than 12 weeks from the acute phase), which highlights the damage caused by the acute phase, as well as the risk of adverse effects and death [17][18]. Leukocytes are responsible for acute dynamics, as well as the production of markers of immunity. Although leukocytes play many different roles against SARS-CoV-2, they are also a key factor in tissue repair and convalescence [19][20]. Higher levels of total leukocytes are described, but the immune cell and soluble protein profile changes during infection (active COVID-19) between mild and severe patients, especially to predict the consequences in convalescence [21][22][23].
2. An Overview of Immunology in the Acute Phase of COVID-19
The establishment of a SARS-CoV-2 infection triggers the host’s immune response to recognition, inflammation, and viral clearance. Previous studies have highlighted the prevalence of neutrophils and monocytes as the first immune cells to migrate to the infectious site through the stimulus of chemokines and the expression of adhesion molecules by endothelial cells, acting as a key factor in innate immunity [24][25]. The recognition of microorganisms and infected cells is mediated by pattern recognition receptors, especially Toll-like receptors (TLRs), NOD-like receptors (NLRs), and others related to intermediate intracellular mechanisms of activation that contribute to the effects observed during the immunological response.
2.1. NETosis Plays a Pivotal Role in COVID-19 Pneumonia Severity
Neutrophils act mainly by phagocytosis, which occurs via the inclusion and digestion of components into intracellular organelles, mediated mainly by enzymes. In addition, neutrophils produce inflammatory mediators, such as reactive oxygen species (ROS), which contribute not only to the activation of other immune cells, but also to cell recruitment to the local site of infection [21][26]. Although cytokines and other proteins have been described in terms of neutrophil interaction in the immune system, their relationship with COVID-19 severity remains scarcely known. Neutrophils play an important role in innate and adaptive responses since it is the first cell to reach the inflammatory site, where they can recognize the pathogen, digest it, and promote an immune response through the release of inflammatory cytokines [27].
The absolute neutrophil count (ANC) has been reported as an important severity mediator among COVID-19 patients, along with lymphocyte count. Patients who required intensive care unit (ICU) admission for pneumonia caused by COVID-19 or who developed the severe form of the disease had higher values of ANC and/or neutrophil-to-lymphocyte-ratio (NLR) [6][10][24][25][28][29][30][31][32][33][34][35]. NLR determination is an easy and cost-effective test to perform in clinical practice, and it has shown a significant improvement in the stratification of COVID-19 patients at hospital admission [21][30][36][37], as well as its potential as a prognostic factor for COVID-19 outcome [34][37][38][39][40], especially in those with comorbidities, such as type 2 diabetes, hypertension, and ischemic heart disease [25][32][41].
Relative values expressed as a percentage of neutrophils, related to absolute leucocyte count, did not demonstrate a significant difference in acute patients, even among individuals with confirmed SARS-CoV-2 infection and those patients exposed [21][42], emerging as a non-recommended parameter in clinical and laboratory COVID-19 management.
The acute phase is marked by inflammatory and inhibitory cell surface markers, such as CD63, CD64, CD117, and CXCR3, driven mainly by CXCL8 and G-CSF [42][43][44][45][46]. The participation of immature neutrophils marked by CD10+ and CD16low is prominent in the severe form, with acute respiratory distress syndrome [28][47] and ICU patients close to discharge, when compared to moderate and mild patients, driven by G-CSF [43]. Due to the urgency of inflammatory mediators, the cell phenotype profile shows a ‘shift to the left’, with the presence of immature neutrophils, although it is unknown whether they are immunosuppressive or pro-inflammatory. A positive correlation of immature neutrophils was seen with inflammatory markers of IL-6, IL-1ra, CXCL8, CXCL10, CCL3, CCL4, and vascular endothelial growth factor [12][43][48][49].
CD11b, another important neutrophil marker related to adhesion to alveolar macrophages [50], was demonstrated to be controversial under COVID-19 disease activity [12][27][51], but was associated with prolonged viral replication, being significantly reduced in those with a poor outcome [51]. From the acute to convalescent stage, this marker was shown not to suffer a significant difference [42].
The expression of activated markers and adhesion molecules is important for a better understanding of neutrophil physiology and further therapeutic strategies. Thus, immature neutrophils demonstrated greater participation in COVID-19 disease, potentially due to their regulatory function, the tissue healing process, and the low expression of adhesion markers, which contribute to their maintenance in peripheral blood [12][49][52].
Severe stages of the disease are characterized by hypoxemia, which was demonstrated to activate transcriptional factor HIF-1α (hypoxia-inducible factor 1α), responsible for further production of an inflammatory profile, guided mainly by IL-1β, IL-6, and IL-8 (CXCL8) [29][53] (Figure 1). Other molecules, such as platelet-derived factor 4 (PF4) and CCL5 chemokine, can trigger neutrophil activation and promote mechanisms that may aggravate the condition of COVID-19 patients [54].
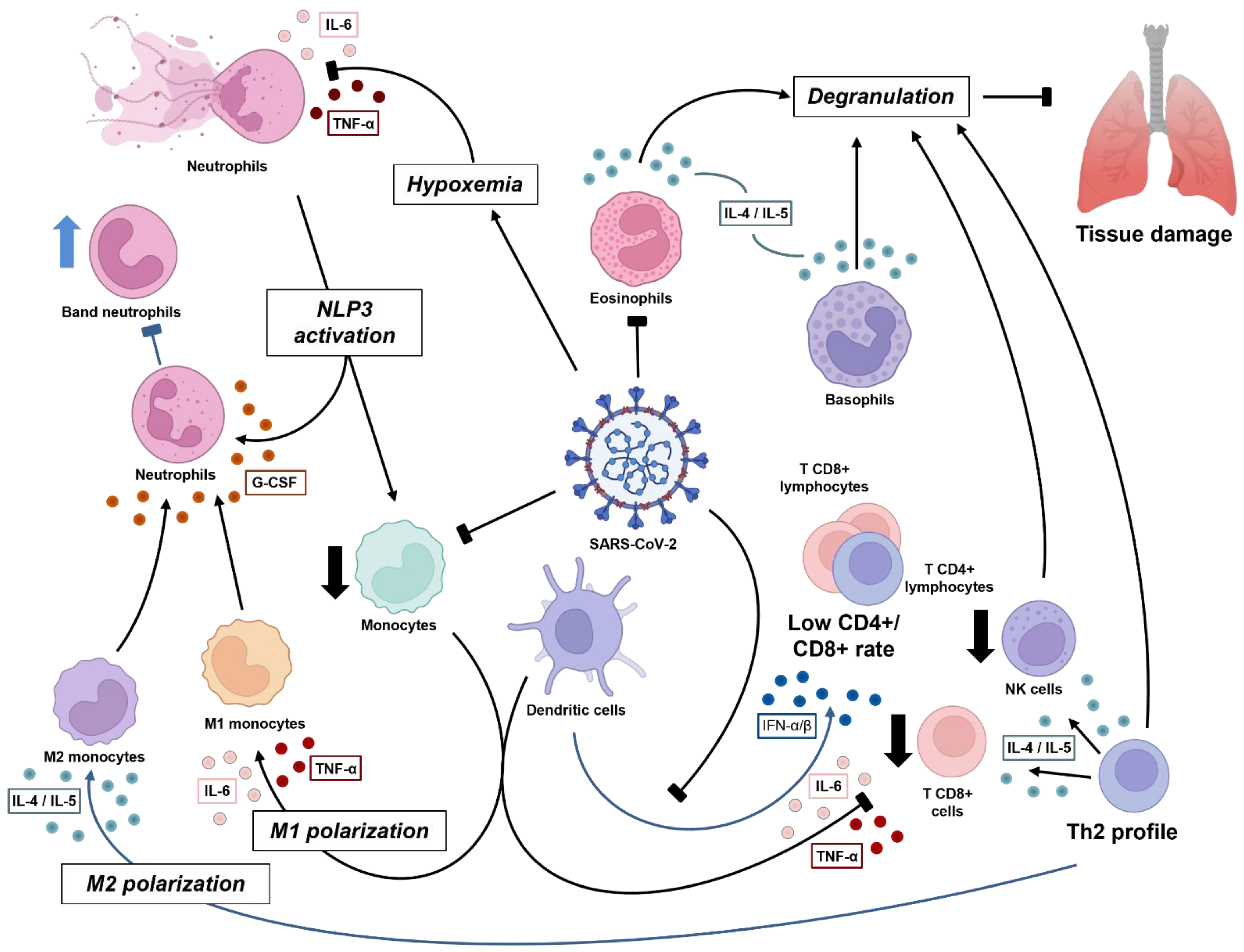
Figure 1. Dynamics of immune cell response against SARS-CoV-2 in acute phase. The virus induces pulmonary damage, which further causes hypoxemia. Both infection and tissue damage lead to inflammatory mediators, mainly interleukin-1β (IL-1β), CXCL8, and IL-6, which further cause NETosis and promote positive inflammatory feedback via inflammasome activation in both neutrophils and monocytes. Subsequently, critical patients experience low monocyte count, in terms of both T CD8+ and natural killer (NK) cells, with M1 polarization (driven by cytokine storm), all related to a worse clinical condition. Instead, when the antiviral stage is established, there is sustained type I interferon (IFN) by dendritic cells, contributing to the Th2 profile (IL-4 and IL-5), which induces the degranulation process by lymphocytes (CD8+ and NK), eosinophils, and basophils, and an M2 polarization, which results in a better prognosis. The release of granules has good and bad effects, whereby they can damage viral and human infected cells, but may also damage normal tissue, characterizing the respiratory syndrome. Macrophages produce granulocyte colony-stimulating factor (G-CSF), which acts in bone marrow to mature leukocyte progenitors. This may lead to a ‘shift to the left’ and improve viral response.
The participation of chemokines and cytokines is still not clear; however, their participation can recruit neutrophils to local activity. When infected by SARS-CoV-2, the lungs express chemokines CXCL1, CXCL2, CXCL3, CXCL5, CXCL8, and CCL20 that induce neutrophil chemotaxis from blood vessels to the lungs [53][55]. Furthermore, cytokines and chemokines in COVID-19 patients are guided mainly by the inflammatory process, involving CCL2, CXCL10, CXCL8, IL-6, and tumor necrosis factor (TNF) [53]. These molecules are important acute inflammatory mediators, which drive chemotaxis, increase adhesion molecules, and induce positive immune regulation from neutrophils and other cells [53].
Novel neutrophil mechanisms have been an important focus of research. Neutrophil extracellular traps (NETs), nets composed mainly of histones and proteins (lactoferrin, cathepsins, elastase, and myeloperoxidase), along with cytoskeleton components and other plasmatic proteins, are among the major mechanisms of neutrophils to maintain homeostasis [56][57]. NET release and ROS production are based on the neutrophil maturation level. Immature granulocytes are reportedly lower during viral clearance, when compared to normal individuals, which suggests that it might reflect the recruitment of mature and efficient neutrophils to leave the circulation and migrate to the tissue [21].
NETs also have the property of activating inflammasome complex NLRP3, which is responsible for type I interferon production and inflammatory cytokines [58]. Additionally, there is a stimulus of adaptive immune response, endothelial injury, thrombosis mediated by the immune system, and occlusion of small vessels, which may worsen pneumonia experienced by COVID-19 patients [43]. On the other hand, the capture of microorganisms and immune cells allows the maintenance of inflammatory damage and downregulates it, thus reducing the consequences of exacerbated inflammation.
Several studies have described the formation of NETs in COVID-19 patients, with increased rates of markers in patients who evolved severe complications, such as thrombosis and death, or in intubated compared to non-intubated patients and convalescents [43][54][57], even though patients with a mild form of COVID-19 express a different type of immune response. NET components myeloperoxidase (MPO)-DNA, neutrophil elastase (NE)-DNA, and citrullinated histones H3 exhibited no significant difference in mild and ICU patients, but they were increased when compared to healthy individuals [12][34][59]. In contrast, higher values of neutrophil elastase were found among ICU patients on entrance, with a median reduction after 7 days and further discharge [43]. Ng et al. [60] suggested a positive correlation of NET production, poor outcome, and inflammatory markers, such as WBC, ANC, and inflammatory cytokines, in addition to increased rates of thrombosis in the COVID-19 group.
A higher concentration of NETs in the respiratory tract than in circulating plasma was also observed, and a direct correlation index to clinical illness severity score was established [54]. It is important to note that NET release induces direct and indirect effects in a systemic manner. Activation of the complement system due to the presence of tissue factor, along with the formation of aggregates in the pulmonary compartment, worsens the clinical condition of pneumonia [56][61].
The thrombosis effects caused by NETs can cause injury not only to the pulmonary tract, but also to the endothelial, renal, liver, and cardiac systems, and they may be involved in further organ failure. Individuals with previous chronic disease, characterized as groups at risk of COVID-19, may experience an increase in endothelial injury due to the intense activity of immune cells, cytokine storm, and inflammatory mediator production [44][54][56]. The participation of thrombotic factors, such as D-dimer and von Willebrand factor, has been reported as being more intense in severe COVID-19 patients; although few studies have described the direct correlation of NETosis and thrombotic molecules, a relationship among NET, platelet aggregates, and the state of thrombosis has already been proposed [44][54][60].
2.2. Improvement of Severe Cases Is Marked by Eosinophilia
Eosinophils are polymorphonuclear cells that play a role in innate immunity; they are characterized by granules extremely rich in inflammatory mediators, such as cationic proteins, peroxidase, hydrolase, and lysophospholipase. This lineage has been extensively studied in terms of parasite response and allergic diseases [62]. This cell profile also presents PRRs and produces cytokines, nitric oxide, and proteins, which contribute to viral clearance [63].
There is an important relationship between cytokines and eosinophil production. It is already known that IL-4 promotes the expression of adhesion molecules that further contribute to eosinophil adhesion to the endothelium, while IL-5 induces degranulation. Both cytokines are produced mainly by mast cells, basophils, and Th2 lymphocytes, which are extremely important in allergic responses. The degranulation process is commonly used in parasites due to membrane damage, but it can also damage the host tissue [62].
Eosinopenia (<40/mm3) was reported in patients admitted to the ICU, where it was suggested as a prognostic factor for poor outcomes [7][8][21][63][64][65][66][67]. In severe patients, CD8+ T cells contribute to eosinophil proliferation via the production of IL-5; however, due to exhaustion on the first cell, the IL-5 level may suffer interference, which might be a cause of eosinopenia at the beginning of the disease course [36]. Around 20 days after hospital admission, the absolute eosinophil count (AEC) usually exceeds 1500/mm3 in patients ready for discharge. This eosinophilia commonly lasts around 5 days and is correlated with a reduced mortality rate [8][53][64][68][69].
Asian race/ethnicity with eosinophilia was a predictor for a shorter hospital stay, while other races/ethnicities showed no significant difference [63]. Other factors that have been proposed to contribute to no resolution of AEC are higher age, alcohol abuse, tobacco use, hypertension, diabetes mellitus, chronic pulmonary disease, chronic kidney failure, comorbidities, previous use of corticoids, and initial symptoms of normal cough, dyspnea, arthromyalgia, asthenia, and saturation >95% [8]. Some studies with asthmatic patients presented the same pattern, in addition to reporting the protective aspect of eosinophilia and a Th2 cytokine profile on disclosure of COVID-19 patients with asthma [63][70].
This cell lineage was also seemingly connected to severe symptoms, with a few patients with normal AEC experiencing fever, fatigue, shortness of breath, and inflammatory infiltrates at hospital admission, in addition to increased rates of aggravation [69], related to the natural killer (NK) T-cell response and further eosinophilic lung inflammation [26].
Sustained eosinopenia was found in severe cases and in patients with cytokine storm syndrome [28][36], and three hypotheses have been suggested [69]: (1) production of corticosteroids by the adrenal during an acute response, which blocks the release of eosinophils from bone marrow and induces migration of eosinophils to the tissue, culminating in reduced eosinophils in circulation; (2) COVID-19 may cause damage to the bone marrow, which would also impact eosinophil count (hypothesis not fully elucidated); (3) upregulation of Th1 and Th2 cytokines by viral clearance promotes leukocyte migration to pulmonary tissue, resulting in a lower availability in peripheral blood. Although not widespread, eosinopenia might also be related to the infection of eosinophils by SARS-CoV-2, as previously demonstrated [9]. These issues can be highlighted, especially as the eosinophil count increases at the same rate as clinical improvement and viral load reduction.
Both eosinophils and neutrophils were found in the bronchoalveolar fluid extracted from patients with severe COVID-19, in addition to eosinophil cationic proteins, which confirms the importance of eosinophils in the local immune response against SARS-CoV-2. The number of eosinophils in the pulmonary tract can cause similar inflammation to acute eosinophilic pneumonia, with a previous observation of >25% of eosinophils in the lungs [26]. Although it is known that COVID-19 is the agent responsible for leukocyte recruitment to the pulmonary tissue, the role of eosinophils in viral clearance is not fully understood.
The surface markers on COVID-19 patients demonstrate an activate profile characterized by a lower expression of CD15, CD66b, and CD193 and a higher expression of CD62L, CD69, and CD147, compared to noninfected individuals. A comparison between moderate and severe patients revealed CD69+ eosinophils in the latter, which might be related to decreased outcome, whereas CD66b, CD11b, CD11a, and CD24 are present in eosinophil membranes in moderate patients, influencing clinical management [28].
Activated eosinophils (CD69+) exhibit a positive correlation with soluble inflammatory molecules in severe patients, such as IFN-γ, CCL2, CCL7, and CCL8. They play a key role in lung tissue infiltration, degranulation of neutrophils, clotting factor activation, molecule recognition, and extracellular matrix metabolization [28].
2.3. Granulocytes and Monocytes Management in Viral Clearance
COVID-19 pathology is guided by a Th2 cytokine profile, which is directly connected to the participation of eosinophils, basophils, and the local inflammatory response. The absolute basophil count (ABC) was found at lower levels during the initial course of the disease, while showing recovery over the course of illness [22]. An absence of ABC recovery was present in severe patients who required mechanical ventilation and who evolved to a fatal outcome, possibly being a biomarker for a poor outcome [51]. A negative relationship between basophil count and both severe and hospitalized COVID-19 patients demonstrates the importance of basophils in local viral clearance [71].
The evaluation of basophils during disease activity demonstrated an increased rate of CD131+ (IL-3 membrane receptor) cells, CD11b, CD63, and CXCR4 [28] but a low expression of GM-CSF and IL-5 receptors [51]. Furthermore, the involvement of thrombotic events in severe cases, as well as immunomodulatory effects during the acute and chronic responses, highlights the important role of basophils in immunity against SARS-CoV-2 [71]. Basophils are involved in the hypersensitivity response, production of mucus, vaso-constriction, inflammation, and tissue damage, but more studies must be conducted to evaluate their effect in acute COVID-19 [62].
Monocytes, on the other hand, represent a subpopulation of leukocytes from the same precursor as neutrophils. They are known as agranulocytes, with a main function related to the recognition of pathogens and cell products by PRRs and subsequent phagocytosis. The stimulus activates intercellular mechanisms that contribute to cytokine storm and migration to tissue.
These cells participate in both innate and adaptive immunity, acting in COVID-19 viral clearance or as antigen-presenting cells to combat the virus or induce antibody production, respectively. Monocytes are classified into three specific subtypes according to their cell surface proteins: classical (CD14++CD16−), inflammatory (CD14++CD16+), and patrolling (CD14+CD16++). It is important to highlight that other markers can also be used, such as chemokine receptors and cytokine production [72].
Monocytes also contribute to an interesting aspect of COVID-19 physiopathology, as they were previously shown to express ACE2, thus being influenced by the virus [73]. It was shown that prolonged viral infection (more than 10 days of positive RT-PCR tests from admission) can reduce ACE2 mRNA, as well as levels of soluble ACE2, instead quickly returning to normal in patients with a negative RT-PCR in less than 10 days [74].
The absolute monocyte count (AMC) is a low-cost measurement, and monocytosis/monocytopenia was previously related to hospital discharge. Those with higher AMC spent fewer days in hospital (15 days), while those with lower levels of AMC remained for a prolonged period (40 days) [73]. In addition to its functionality, low rates of monocytes were found in severe and in mechanical ventilation patients [21][40][51][75], further associated with age [33] and the presence of atypical and vacuolated monocytes [73]. It was suggested that these morphological changes come from the process of monocyte infection, as also observed in visceral leishmaniasis, but not other viral diseases [73].
Severe disease is marked by a lower rate of AMC when compared to mild cases [40][75][76]. Monocyte soluble markers show that, during acute COVID-19, there is a higher secretion of sCD14 and sCD163 when compared to normal individuals. sCD163 is correlated with the time elapsed from hospital admission, whereas sCD14 is correlated with several laboratory parameters, including IL-6 and C reactive protein (CRP). Accordingly, a few differences were observed between ICU and non-ICU patients, but patrolling monocytes produce less sCD163 and more sCD14 [23][49][77][78][79]. Corticoids were suggested as a factor interfering with monocyte activation and inflammatory pathways, although the CD163 receptor was increased in all subtypes of monocytes in severe conditions [10][75][80][81][82].
The participation of monocytes/macrophages in pulmonary inflammatory diseases has been reported, especially classical monocytes in asthma [83][84]. Some studies have reported higher levels of monocytes in peripheral blood with a further reduction in convalescent stage, whereas others have reported the opposite [21][85][86], with similar observations in bronchoalveolar fluid [26]. A delay in interferon signaling leads to the infiltration of monocytes into the pulmonary tract, thus inducing the production of inflammatory mediators that drive the response to cytokine storm, which results in positive feedback to leukocytes, contributing to tissue damage and regulation of antiviral cytokines. Coronaviruses mediate antiviral cytokines via translational mechanisms that are usually involved in viral clearance, such as type I interferon, which acts as an escape mechanism [87].
During cytokine storm syndrome, monocytes tend to reduce their quantitative circulating value, but increase the granularity and permeability of endothelial cells [36][85]. This might result in the expression of adhesion molecules and migration to other tissues. Although it is well established that cytokine storm is the main event that influences a worse prognosis, monocytes (and even dendritic cells) were suggested to not be the main producers of proinflammatory cytokines [88].
The infection stage involves the participation of CD16+ monocytes, in contrast to a healthy status [73][79]. However, among COVID-19 subgroups, mild and severe stages presented increased rates of inflammation (CD16+) and presenting ability (HLA-DR+) when compared to critical patients [49][51][75][76][81]. Winheim et al. [89] demonstrated that, within the subpopulation of activated monocytes, there was participation mainly by classical monocytes. This inflammatory profile was demonstrated to be guided by higher levels of TNF-α, IL-6, IL-10, IL-2, IL-4, IL-13, IL-18, CCL3, CCL4, and CCL2, although only IL-6 had a significant positive relationship with CD16+ monocytes and a negative relationship with HLA-DR+ monocytes [19][76][90][91][92][93]. This suggests that proinflammatory monocytes drive IL-6 production, which may be related to the proinflammatory state and low HLA-DR production in critical patients (Figure 1).
Researchers must highlight that both extra- and intracellular mechanisms play an important role in SARS-CoV-2 clearance, especially inflammasome activation, as mentioned before, in neutrophils and monocytes. Interactions among viral RNA [94], NETs [43][58], and the dysregulation of calcium concentration [95] influence NLRP3 activation, mediated by the viral envelope protein. Upon stimulus, this induces the maturation of proinflammatory cytokines IL-1β and IL-18, which further stimulates IL-6 and TNF, promoting inflammation in the lungs. These events worsen disease progression, representing the major mediators of cytokine storm, and cause systemic dysfunctions, such as macrophage recruitment and leukocyte degranulation [96]. Moreover, severe patients experience an increased rate of activation of the NLRP3 and TXNIP inflammasome pathways [79].
The expression of surface markers demonstrates a predominance of M1 macrophages in critical patients, when compared to noncritical patients, due to the increased expression of CD80, higher production of IL-6, TNF-α, and TGF-β cytokines (although M1 macrophages are not the main source of these cytokines), and lower expression of MHC-II [73][82][88][97]. It is important to note that M1 macrophages show increased odds of tissue migration due to their rheologic properties, but also fewer acid granules, which contribute to viral RNA perseverance in the cell. M2 macrophages, otherwise, present more acid granules, which lead to viral RNA instability and further degradation [90][98]. Even though alveolar M2 macrophages (CD206+) are also present during the immune response, when compared to the healthy status, the cytokine profile barely varied, with an IL-4/IL-13 balance to induce M2 macrophages [73][99]; however, a decrease in CD86+ expression and MHC-II [88] was observed.
Dendritic cells (DCs) play an important role in viral clearance, antigen presentation from innate to adaptive immunity, and the capture of apoptotic/necrotic cells [100]. Immature DCs show an increased ability to recognize antigens, while mature DCs are important producers of IL-12, IL-1β, type I and type II IFN, IL-4, IL-10, and TNF-α. [101]. Their function in COVID-19 disease remains unclear, and their participation is mainly guided by inflammatory status. Some studies demonstrated a lower expression of c-KIT+ in cDC1 and of plasmacytoid DCs in mild/moderate patients, together with a significant reduction in DC count. Moderate patients were marked by a higher expression of CD38. However, with the progression of disease severity, a lower participation of the inflammatory DC3 subset (CD163−CD14−) was reported, with an increase in the c-KIT receptor [49][81][102][103].
Patients both with and without neurological symptoms 4 weeks after disease onset demonstrate increased rates of DC density and mature DCs, when compared to a healthy status. However, differences were not observed between infected groups [104]. These data suggest long-term participation of DCs, whether the patient is symptomatic or not. Cell activation and inflammatory status are compromised by increased age, and senescence is a common factor of immunity, which, when related to COVID-19, seems to play a key role in response, potentially reflecting why newer patients experience disease differently [101].
During viral infection, plasmacytoid DCs (pDC) are important producers of type I IFN (especially IFN-α) via recognition of RNA by TLR7/8, once there is participation of the PD-L1+CD80− DC population; however, their levels are diminished with severity [49]. Even though a phenotypic profile is prominent in asymptomatic patients, hospitalized patients display a phenotype characterized by PD-L1+CD80+. It is important to highlight the higher expression of CD86 in asymptomatic patients [49], demonstrating their greater ability to stimulate DCs, whereas hospitalized patients had a higher expression of CD80 [12][103] and lower HLA-DR [105].
During viral infections (including viruses other than coronavirus), viral escape can occur through interference with type I IFN production via antagonism of transcriptional factors. This was established by some studies describing a higher concentration of IFN-α and IFN-genes at the beginning of disease, with a subsequent reduction, even in patients that experience a severe form, in comparison to mild patients [12][106][107]. IFN-α plays an important role in dendritic cell functionality during acute disease, even in convalescence. A normal level is not restored for more than 6 months after infection, and a positive correlation index between IFN-α cytokine and pDCs has been demonstrated, which might be related to the decrease in P1-pDCs in hospitalized patients [106]. Costimulatory molecules CD80 (B7-1) and CD86 (B7-2), expressed mainly by DCs, are capable of activating T cells and participating in the SARS-CoV-2 response. A lower circulating level of mDC and pDC CD86+ has been reported in the acute phase; however, their efficiency in activating T lymphocytes must be explored, as their dynamics in acute disease interfere with antibody titers during convalescence [106][108].
References
- Dousari, A.S.; Moghadam, M.T.; Satarzadeh, N. COVID-19 (Coronavirus Disease 2019): A New Coronavirus Disease. Infect. Drus Resist. 2020, 13, 2819–2828.
- Khan, A.H.; Tirth, V.; Fawzy, M.; Mahmoud, A.E.D.; Khan, N.A.; Ahmed, S.; Ali, S.S.; Akram, M.; Hameed, L.; Islam, S.; et al. COVID-19 Transmission, Vulnerability, Persistence and Nanotherapy: A Review. Environ. Chem. Lett. 2021, 19, 2773–2787.
- Zhang, Y.-Z.; Holmes, E.C. A Genomic Perspective on the Origin and Emergence of SARS-CoV-2. Cell 2020, 181, 223–227.
- Cui, J.; Li, F.; Shi, Z.-L. Origin and Evolution of Pathogenic Coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192.
- Taghiloo, S.; Aliyali, M.; Abedi, S.; Mehravaran, H.; Sharifpour, A.; Zaboli, E.; Eslami-Jouybari, M.; Ghasemian, R.; Vahedi-Larijani, L.; Hossein-Nattaj, H.; et al. Apoptosis and Immunophenotyping of Peripheral Blood Lymphocytes in Iranian COVID-19 Patients: Clinical and Laboratory Characteristics. J. Med. Virol. 2021, 93, 1589–1598.
- Balzanelli, M.G.; Distratis, P.; Dipalma, G.; Vimercati, L.; Catucci, O.; Amatulli, F.; Cefalo, A.; Lazzaro, R.; Palazzo, D.; Aityan, S.K.; et al. Immunity Profiling of COVID-19 Infection, Dynamic Variations of Lymphocyte Subsets, a Comparative Analysis on Four Different Groups. Microorganisms 2021, 9, 2036.
- Liu, H.; Chen, J.; Yang, Q.; Lei, F.; Zhang, C.; Qin, J.-J.; Wang, Y.; Cai, J.; Li, H. Development and Validation of a Risk Score Using Complete Blood Count to Predict In-Hospital Mortality in COVID-19 Patients. Med 2021, 2, 435–447.
- González, M.M.; Gonzalo, E.S.; Lopez, I.C.; Fernández, F.A.; Pérez, J.L.B.; Monge, D.M.; Núñez, J.A.V.; Fenoll, R.G.; Fernández, C.S.; Castro, S.J.F.; et al. The Prognostic Value of Eosinophil Recovery in COVID-19: A Multicentre, Retrospective Cohort Study on Patients Hospitalised in Spanish Hospitals. J. Clin. Med. 2021, 10, 305.
- Wei, Y.; Lu, Y.; Xia, L.; Yuan, X.; Li, G.; Li, X.; Liu, L.; Liu, W.; Zhou, P.; Wang, C.Y.; et al. Analysis of 2019 Novel Coronavirus Infection and Clinical Characteristics of Outpatients: An Epidemiological Study from a Fever Clinic in Wuhan, China. J. Med. Virol. 2020, 92, 2758–2767.
- Zhao, G.; Su, Y.; Sun, X.; Cui, X.; Dang, L.; Zhao, L.; Tan, X.; Wang, H.; Yang, M. A Comparative Study of the Laboratory Features of COVID-19 and Other Viral Pneumonias in the Recovery Stage. J. Clin. Lab. Anal. 2020, 34, e23483.
- Jafarzadeh, A.; Chauhan, P.; Saha, B.; Jafarzadeh, S.; Nemati, M. Contribution of Monocytes and Macrophages to the Local Tissue Inflammation and Cytokine Storm in COVID-19: Lessons from SARS and MERS, and Potential Therapeutic Interventions. Life Sci. 2020, 257, 118102.
- Parackova, Z.; Zentsova, I.; Bloomfield, M.; Vrabcova, P.; Smetanova, J.; Klocperk, A.; Mesežnikov, G.; Fernando, L.; Mendez, C.; Vymazal, T.; et al. Disharmonic Inflammatory Signatures in COVID-19: Augmented Neutrophils’ but Impaired Monocytes’ and Dendritic Cells’ Responsiveness. Cells 2020, 9, 2206.
- Garanina, E.; Hamza, S.; Stott-Marshall, R.J.; Martynova, E.; Markelova, M.; Davidyuk, Y.; Shakirova, V.; Kaushal, N.; Baranwal, M.; Khaertynova, I.M.; et al. Antibody and T Cell Immune Responses to SARS-CoV-2 Peptides in COVID-19 Convalescent Patients. Front. Microbiol. 2022, 13, 842232.
- Zhao, P.; Zou, J.; Zhou, F.; Zhu, Y.; Song, Q.; Yu, D.; Li, X. Immune Features of COVID-19 Convalescent Individuals Revealed by a Single-Cell RNA Sequencing. Int. Immunopharmacol. 2022, 108, 108767.
- Zhou, Y.; Zhang, J.; Wang, D.; Wang, D.; Guan, W.; Qin, J.; Xu, X.; Fang, J.; Fu, B.; Zheng, X.; et al. Profiling of the Immune Repertoire in COVID-19 Patients with Mild, Severe, Convalescent, or Retesting-Positive Status. J. Autoimmun. 2021, 118, 102596.
- Pan, Y.; Jiang, X.; Yang, L.; Chen, L.; Zeng, X.; Liu, G.; Tang, Y.; Qian, C.; Wang, X.; Cheng, F.; et al. SARS-CoV-2-Specific Immune Response in COVID-19 Convalescent Individuals. Signal Transduct. Target. Ther. 2021, 6, 256.
- Yan, Z.; Yang, M.; Lai, C.L. Long COVID-19 Syndrome: A Comprehensive Review of Its Effect on Various Organ Systems and Recommendation on Rehabilitation Plans. Biomedicines 2021, 9, 966.
- Fernández-de-las-Peñas, C.; Palacios-Ceña, D.; Gómez-Mayordomo, V.; Cuadrado, M.L.; Florencio, L.L. Defining Post-COVID Symptoms (Post-Acute COVID, Long COVID, Persistent Post-COVID): An Integrative Classification. Int. J. Environ. Res. Public Health 2021, 18, 2621.
- Kumar, P.; Sah, A.K.; Tripathi, G.; Kashyap, A.; Tripathi, A.; Rao, R.; Mishra, P.C.; Mallick, K.; Husain, A.; Kashyap, M.K. Role of ACE2 Receptor and the Landscape of Treatment Options from Convalescent Plasma Therapy to the Drug Repurposing in COVID-19. Mol. Cell Biochem. 2021, 476, 553–574.
- Azkur, A.K.; Akdis, M.; Azkur, D.; Sokolowska, M.; van de Veen, W.; Brüggen, M.C.; O’Mahony, L.; Gao, Y.; Nadeau, K.; Akdis, C.A. Immune Response to SARS-CoV-2 and Mechanisms of Immunopathological Changes in COVID-19. Allergy Eur. J. Allergy Clin. Immunol. 2020, 75, 1564–1581.
- Kwiecień, I.; Rutkowska, E.; Kulik, K.; Kłos, K.; Plewka, K.; Raniszewska, A.; Rzepecki, P.; Chciałowski, A. Neutrophil Maturation, Reactivity and Granularity Research Parameters to Characterize and Differentiate Convalescent Patients from Active SARS-CoV-2 Infection. Cells 2021, 10, 2332.
- Mao, J.; Dai, R.; Du, R.-C.; Zhu, Y.; Shui, L.-P.; Luo, X.-H. Hematologic Changes Predict Clinical Outcome in Recovered Patients with COVID-19. Ann. Hematol. 2021, 100, 675–689.
- Hasan, M.Z.; Islam, S.; Matsumoto, K.; Kawai, T. Meta-Analysis of Single-Cell RNA-Seq Data Reveals Phenotypic Switching of Immune Cells in Severe COVID-19 Patients. Comput. Biol. Med. 2021, 137, 104792.
- Zhang, B.; Zhou, X.; Zhu, C.; Song, Y.; Feng, F.; Qiu, Y.; Feng, J.; Jia, Q.; Song, Q.; Zhu, B.; et al. Immune Phenotyping Based on the Neutrophil-to-Lymphocyte Ratio and IgG Level Predicts Disease Severity and Outcome for Patients With COVID-19. Front. Mol. Biosci. 2020, 7, 157.
- Moradi, E.V.; Teimouri, A.; Rezaee, R.; Morovatdar, N.; Foroughian, M.; Layegh, P.; Kakhki, B.R.; Koupaei, S.R.A.; Ghorani, V. Increased Age, Neutrophil-to-Lymphocyte Ratio (NLR) and White Blood Cells Count Are Associated with Higher COVID-19 Mortality. Am. J. Emerg. Med. 2021, 40, 11–14.
- Kim, D.-M.; Seo, J.-W.; Kim, Y.; Park, U.; Ha, N.-Y.; Park, H.; Yun, N.R.; Kim, D.Y.; Yoon, S.H.; Na, Y.S.; et al. Eosinophil-Mediated Lung Inflammation Associated with Elevated Natural Killer T Cell Response in COVID-19 Patients. Korean J. Intern. Med. 2022, 37, 201–209.
- Chao, Y.; Rebetz, J.; Bläckberg, A.; Hovold, G.; Sunnerhagen, T.; Rasmussen, M.; Semple, J.W.; Shannon, O. Distinct Phenotypes of Platelet, Monocyte, and Neutrophil Activation Occur during the Acute and Convalescent Phase of COVID-19. Platelets 2021, 32, 1092–1102.
- Lourda, M.; Dzidic, M.; Hertwig, L.; Bergsten, H.; Palma Medina, L.M.; Sinha, I.; Kvedaraite, E.; Chen, P.; Muvva, J.R.; Gorin, J.-B.; et al. High-Dimensional Profiling Reveals Phenotypic Heterogeneity and Disease-Specific Alterations of Granulocytes in COVID-19. Proc. Natl. Acad. Sci. USA 2021, 118, e2109123118.
- McElvaney, O.J.; McEvoy, N.L.; McElvaney, O.F.; Carroll, T.P.; Murphy, M.P.; Dunlea, D.M.; Choileáin, O.N.; Clarke, J.; O’Connor, E.; Hogan, G.; et al. Characterization of the Inflammatory Response to Severe COVID-19 Illness. Am. J. Respir. Crit. Care Med. 2020, 202, 812–821.
- Cai, J.; Li, H.; Zhang, C.; Chen, Z.; Liu, H.; Lei, F.; Qin, J.; Liu, Y.; Zhou, F.; Song, X.; et al. The Neutrophil-to-Lymphocyte Ratio Determines Clinical Efficacy of Corticosteroid Therapy in Patients with COVID-19. Cell Metab. 2021, 33, 258–269.
- Ducastel, M.; Chenevier-Gobeaux, C.; Ballaa, Y.; Meritet, J.F.; Brack, M.; Chapuis, N.; Pene, F.; Carlier, N.; Szwebel, T.A.; Roche, N.; et al. Oxidative Stress and Inflammatory Biomarkers for the Prediction of Severity and ICU Admission in Unselected Patients Hospitalized with COVID-19. Int. J. Mol. Sci. 2021, 22, 7462.
- Imran, M.M.; Ahmad, U.; Usman, U.; Ali, M.; Shaukat, A.; Gul, N. Neutrophil/Lymphocyte Ratio—A Marker of COVID-19 Pneumonia Severity. Int. J. Clin. Pract. 2020, 75, e13698.
- Lanini, S.; Montaldo, C.; Nicastri, E.; Vairo, F.; Agrati, C.; Petrosillo, N.; Scognamiglio, P.; Antinori, A.; Puro, V.; Di Caro, A.; et al. COVID-19 Disease-Temporal Analyses of Complete Blood Count Parameters over Course of Illness, and Relationship to Patient Demographics and Management Outcomes in Survivors and Non-Survivors: A Longitudinal Descriptive Cohort Study. PLoS ONE 2020, 15, e0244129.
- Leppkes, M.; Knopf, J.; Naschberger, E.; Lindemann, A.; Singh, J.; Herrmann, I.; Stürzl, M.; Staats, L.; Mahajan, A.; Schauer, C.; et al. Vascular Occlusion by Neutrophil Extracellular Traps in COVID-19. EBioMedicine 2020, 58, 102925.
- Liu, G.; Zhang, S.; Hu, H.; Liu, T.; Huang, J. The Role of Neutrophil-Lymphocyte Ratio and Lymphocyte–Monocyte Ratio in the Prognosis of Type 2 Diabetics with COVID-19. Scott. Med. J. 2020, 65, 154–160.
- Martens, R.J.H.; Van Adrichem, A.J.; Mattheij, N.J.A.; Brouwer, C.G.; Van Twist, D.J.L.; Broerse, J.J.C.R.; Magro-Checa, C.; Van Dongen, C.M.P.; Mostard, R.L.M.; Ramiro, S.; et al. Hemocytometric Characteristics of COVID-19 Patients with and without Cytokine Storm Syndrome on the Sysmex XN-10 Hematology Analyzer. Clin. Chem. Lab. Med. 2021, 59, 783–793.
- Taj, S.; Kashif, A.; Fatima, S.A.; Imran, S.; Lone, A.; Ahmed, Q. Role of Hematological Parameters in the Stratification of COVID-19 Disease Severity. Ann. Med. Surg. 2021, 62, 68–72.
- Man, M.A.; Rajnoveanu, R.M.; Motoc, N.S.; Bondor, C.I.; Chis, A.F.; Lesan, A.; Puiu, R.; Lucaciu, S.R.; Dantes, E.; Gergely-Domokos, B.; et al. Neutrophil-to-Lymphocyte Ratio, Platelets-to-Lymphocyte Ratio, and Eosinophils Correlation with High-Resolution Computer Tomography Severity Score in COVID-19 Patients. PLoS ONE 2021, 16, e0252599.
- Rodriguez, L.; Pekkarinen, P.T.; Lakshmikanth, T.; Tan, Z.; Consiglio, C.R.; Pou, C.; Chen, Y.; Mugabo, C.H.; Nguyen, N.A.; Nowlan, K.; et al. Systems-Level Immunomonitoring from Acute to Recovery Phase of Severe COVID-19. Cell Rep. Med. 2020, 1, 100078.
- Asghar, M.S.; Khan, N.A.; Kazmi, S.J.H.; Ahmed, A.; Hassan, M.; Jawed, R.; Akram, M.; Memon, G.M.; Ahmed, M.U.; Tirmizi, S.B.; et al. Hematological Parameters Predicting Severity and Mortality in COVID-19 Patients of Pakistan: A Retrospective Comparative Analysis. J. Community Hosp. Intern. Med. Perspect. 2020, 10, 514–520.
- Liu, J.; Li, S.; Liu, J.; Liang, B.; Wang, X.; Wang, H.; Han, C.; Zhu, H.; Zhou, R.; Zhou, H.; et al. Longitudinal Characteristics of Lymphocyte Responses and Cytokine pro Fi Les in the Peripheral Blood of SARS-CoV-2 Infected Patients. EBioMedicine 2020, 55, 102763.
- Neeland, M.R.; Bannister, S.; Clifford, V.; Dohle, K.; Mulholland, K.; Sutton, P.; Curtis, N.; Steer, A.C.; Burgner, D.P.; Crawford, N.W.; et al. Innate Cell Profiles during the Acute and Convalescent Phase of SARS-CoV-2 Infection in Children. Nat. Commun. 2021, 12, 1084.
- Metzemaekers, M.; Cambier, S.; Blanter, M.; Vandooren, J.; Carvalho, A.C.d.; Malengier-Devlies, B.; Vanderbeke, L.; Jacobs, C.; Coenen, S.; Martens, E.; et al. Kinetics of Peripheral Blood Neutrophils in Severe Coronavirus Disease 2019. Clin. Transl. Immunol. 2021, 10, e1271.
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Weinberger, T.; Weigand, M.; Muenchhoff, M.; Hellmuth, J.C.; Ledderose, S.; Schulz, H.; et al. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated with Respiratory Failure and Coagulopathy. Circulation 2020, 142, 1176–1189.
- Seery, V.; Raiden, S.C.; Algieri, S.C.; Grisolía, N.A.; Filippo, D.; De Carli, N.; Di Lalla, S.; Cairoli, H.; Chiolo, M.J.; Meregalli, C.N.; et al. Blood Neutrophils from Children with COVID-19 Exhibit Both Inflammatory and Anti-Inflammatory Markers. EBioMedicine 2021, 67, 103357.
- Caldarale, F.; Giacomelli, M.; Garrafa, E.; Tamassia, N.; Morreale, A.; Poli, P.; Timpano, S.; Baresi, G.; Zunica, F.; Cattalini, M.; et al. Plasmacytoid Dendritic Cells Depletion and Elevation of IFN-γ Dependent Chemokines CXCL9 and CXCL10 in Children with Multisystem Inflammatory Syndrome. Front. Immunol. 2021, 12, 654587.
- Reyes, L.; Sanchez-Garcia, M.A.; Morrison, T.; Howden, A.J.M.; Watts, E.R.; Arienti, S.; Sadiku, P.; Coelho, P.; Mirchandani, A.S.; Zhang, A.; et al. A Type I IFN, Prothrombotic Hyperinflammatory Neutrophil Signature Is Distinct for COVID-19 ARDS. Wellcome Open Res. 2021, 6, 38.
- Wilk, A.J.; Rustagi, A.; Zhao, N.Q.; Roque, J.; Martínez-Colón, G.J.; McKechnie, J.L.; Ivison, G.T.; Ranganath, T.; Vergara, R.; Hollis, T.; et al. A Single-Cell Atlas of the Peripheral Immune Response in Patients with Severe COVID-19. Nat. Med. 2020, 26, 1070–1076.
- Carissimo, G.; Xu, W.; Kwok, I.; Abdad, M.Y.; Chan, Y.H.; Fong, S.W.; Puan, K.J.; Lee, C.Y.P.; Yeo, N.K.W.; Amrun, S.N.; et al. Whole Blood Immunophenotyping Uncovers Immature Neutrophil-to-VD2 T-Cell Ratio as an Early Marker for Severe COVID-19. Nat. Commun. 2020, 11, 5243.
- Duan, M.; Steinfort, D.P.; Smallwood, D.; Hew, M.; Chen, W.; Ernst, M.; Irving, L.B.; Anderson, G.P. CD11b Immunophenotyping Identifies Inflammatory Profiles in the Mouse and Human Lungs. Mucosal Immunol. 2016, 9, 550–563.
- Renner, K.; Schwittay, T.; Chaabane, S.; Gottschling, J.; Müller, C.; Tiefenböck, C.; Salewski, J.N.; Winter, F.; Buchtler, S.; Balam, S.; et al. Severe T Cell Hyporeactivity in Ventilated COVID-19 Patients Correlates with Prolonged Virus Persistence and Poor Outcomes. Nat. Commun. 2021, 12, 3006.
- Deschler, S.; Kager, J.; Erber, J.; Fricke, L.; Koyumdzhieva, P.; Georgieva, A.; Lahmer, T.; Wiessner, J.R.; Voit, F.; Schneider, J.; et al. Mucosal-Associated Invariant T (MAIT) Cells Are Highly Activated and Functionally Impaired in COVID-19 Patients. Viruses 2021, 13, 241.
- Gebremeskel, S.; Schanin, J.; Coyle, K.M.; Butuci, M.; Luu, T.; Brock, E.C.; Xu, A.; Wong, A.; Leung, J.; Korver, W.; et al. Mast Cell and Eosinophil Activation Are Associated with COVID-19 and TLR-Mediated Viral Inflammation: Implications for an Anti-Siglec-8 Antibody. Front. Immunol. 2021, 12, 650331.
- Middleton, E.A.; He, X.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil Extracellular Traps Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood 2020, 136, 1169–1179.
- Didangelos, A. Neutrophil Involvement in COVID-19. Preprints 2020, 12, 652470.
- Ackermann, M.; Anders, H.J.; Bilyy, R.; Bowlin, G.L.; Daniel, C.; De Lorenzo, R.; Egeblad, M.; Henneck, T.; Hidalgo, A.; Hoffmann, M.; et al. Patients with COVID-19: In the Dark-NETs of Neutrophils. Cell Death Differ. 2021, 28, 3125–3139.
- Zuo, Y.; Zuo, M.; Yalavarthi, S.; Gockman, K.; Madison, J.A.; Shi, H.; Woodard, W.; Lezak, S.P.; Lugogo, N.L.; Knight, J.S.; et al. Neutrophil Extracellular Traps and Thrombosis in COVID-19. J. Thromb. Thrombolysis 2021, 51, 446–453.
- Aymonnier, K.; Ng, J.; Fredenburgh, L.E.; Zambrano-Vera, K.; Munzer, P.; Gutch, S.; Fukui, S.; Desjardins, M.; Subramaniam, M.; Baron, R.M.; et al. Inflammasome Activation in Neutrophils of Patients with Severe COVID-19. Blood Adv. 2022, 6, 2001–2013.
- Arcanjo, A.; Logullo, J.; Menezes, C.C.B.; de Souza Carvalho Giangiarulo, T.C.; dos Reis, M.C.; de Castro, G.M.M.; da Silva Fontes, Y.; Todeschini, A.R.; Freire-de-Lima, L.; Decoté-Ricardo, D.; et al. The Emerging Role of Neutrophil Extracellular Traps in Severe Acute Respiratory Syndrome Coronavirus 2 (COVID-19). Sci. Rep. 2020, 10, 19630.
- Ng, H.; Havervall, S.; Rosell, A.; Aguilera, K.; Parv, K.; Von Meijenfeldt, F.A.; Lisman, T.; Mackman, N.; Thålin, C.; Phillipson, M. Circulating Markers of Neutrophil Extracellular Traps Are of Prognostic Value in Patients with COVID-19. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 988–994.
- Zuo, Y.; Kanthi, Y.; Knight, J.S.; Kim, A.H.J. The Interplay between Neutrophils, Complement, and Microthrombi in COVID-19. Best Pract. Res. Clin. Rheumatol. 2021, 35, 101661.
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Cellular and Molecular Immunology; Elsevier: Philadelphia, PA, USA, 2018; Volume 9.
- Glickman, J.W.; Pavel, A.B.; Guttman-Yassky, E.; Milller, R.L. The Role of Circulating Eosinophils on COVID-19 Mortality Varies by Race/Ethnicity. Allergy 2021, 76, 925–927.
- Georgakopoulou, V.E.; Garmpis, N.; Damaskos, C.; Valsami, S.; Dimitroulis, D.; Diamantis, E.; Farmaki, P.; Papageorgiou, C.V.; Makrodimitri, S.; Gravvanis, N.; et al. The Impact of Peripheral Eosinophil Counts and Eosinophil to Lymphocyte Ratio (ELR) in the Clinical Course of COVID-19 Patients: A Retrospective Study. In Vivo 2021, 35, 641–648.
- Tan, Y.; Zhou, J.; Zhou, Q.; Hu, L.; Long, Y. Role of Eosinophils in the Diagnosis and Prognostic Evaluation of COVID-19. J. Med. Virol. 2021, 93, 1105–1110.
- Vitte, J.; Diallo, A.B.; Boumaza, A.; Lopez, A.; Michel, M.; Allardet-Servent, J.; Mezouar, S.; Sereme, Y.; Busnel, J.M.; Miloud, T.; et al. A Granulocytic Signature Identifies COVID-19 and Its Severity. J. Infect. Dis. 2020, 222, 1985–1996.
- Yan, B.; Yang, J.; Xie, Y.; Tang, X. Relationship between Blood Eosinophil Levels and COVID-19 Mortality. World Allergy Organ. J. 2021, 14, 100521.
- Fraissé, M.; Logre, E.; Mentec, H.; Cally, R.; Plantefève, G.; Contou, D. Eosinophilia in Critically Ill COVID-19 Patients: A French Monocenter Retrospective Study. Crit. Care 2020, 24, 635.
- Xie, G.; Ding, F.; Han, L.; Yin, D.; Lu, H.; Zhang, M. The Role of Peripheral Blood Eosinophil Counts in COVID-19 Patients. Allergy Eur. J. Allergy Clin. Immunol. 2021, 76, 471–482.
- Ferastraoaru, D.; Hudes, G.; Jerschow, E.; Jariwala, S.; Karagic, M.; de Vos, G.; Rosenstreich, D.; Ramesh, M. Eosinophilia in Asthma Patients Is Protective Against Severe COVID-19 Illness. J. Allergy Clin. Immunol. Pract. 2021, 9, 1152–1162.e3.
- Sun, Y.; Zhou, J.; Ye, K. White Blood Cells and Severe COVID-19: A Mendelian Randomization Study. J. Pers. Med. 2021, 11, 195.
- Antonelli, L.R.V.; Leoratti, F.M.S.; Costa, P.A.C.; Rocha, B.C.; Diniz, S.Q.; Tada, M.S.; Pereira, D.B.; Teixeira-Carvalho, A.; Golenbock, D.T.; Gonçalves, R.; et al. The CD14+CD16+ Inflammatory Monocyte Subset Displays Increased Mitochondrial Activity and Effector Function During Acute Plasmodium Vivax Malaria. PLoS Pathog. 2014, 10, e1004393.
- Zhang, D.; Guo, R.; Liu, H.; Wang, Y.; Wang, Y.; Qian, H.; Dai, T.; Zhang, T.; Lai, Y.; Wang, J.; et al. COVID-19 Infection Induces Readily Detectable Morphologic and Inflammation-Related Phenotypic Changes in Peripheral Blood Monocytes. J. Leukoc. Biol. 2020, 1, 13–22.
- Osman, I.O.; Melenotte, C.; Brouqui, P.; Million, M.; Lagier, J.C.; Parola, P.; Stein, A.; La Scola, B.; Meddeb, L.; Mege, J.L.; et al. Expression of ACE2, Soluble ACE2, Angiotensin I, Angiotensin II and Angiotensin-(1-7) Is Modulated in COVID-19 Patients. Front. Immunol. 2021, 12, 625732.
- Rajamanickam, A.; Kumar, N.P.; Pandiarajan, A.N.; Selvaraj, N.; Munisankar, S.; Renji, R.M.; Venkatramani, V.; Murhekar, M.; Thangaraj, J.W.V.; Kumar, M.S.; et al. Dynamic Alterations in Monocyte Numbers, Subset Frequencies and Activation Markers in Acute and Convalescent COVID-19 Individuals. Sci. Rep. 2021, 11, 20254.
- Qin, S.; Jiang, Y.; Wei, X.; Liu, X.; Guan, J.; Chen, Y.; Lu, H.; Qian, J.; Wang, Z.; Lin, X. Dynamic Changes in Monocytes Subsets in COVID-19 Patients. Hum. Immunol. 2021, 82, 170–176.
- Zingaropoli, M.A.; Nijhawan, P.; Carraro, A.; Pasculli, P. Increased SCD163 and SCD14 Plasmatic Levels and Depletion of Peripheral Blood Pro-Inflammatory Monocytes, Myeloid and Plasmacytoid Dendritic Cells in Patients with Severe COVID-19 Pneumonia. Front. Immunol. 2021, 12, 627548.
- Lage, S.L.; Amaral, E.P.; Hilligan, K.L.; Laidlaw, E.; Rupert, A.; Namasivayan, S.; Rocco, J.; Galindo, F.; Kellogg, A.; Kumar, P.; et al. Persistent Oxidative Stress and Inflammasome Activation in CD14highCD16−Monocytes from COVID-19 Patients. Front. Immunol. 2022, 12, 799558.
- Flament, H.; Rouland, M.; Beaudoin, L.; Toubal, A.; Bertrand, L.; Lebourgeois, S.; Rousseau, C.; Soulard, P.; Gouda, Z.; Cagninacci, L.; et al. Outcome of SARS-CoV-2 Infection Is Linked to MAIT Cell Activation and Cytotoxicity. Nat. Immunol. 2021, 22, 322–335.
- Gómez-Rial, J.; Rivero-Calle, I.; Salas, A.; Martinón-Torres, F. Role of Monocytes/Macrophages in COVID-19 Pathogenesis: Implications for Therapy. Infect. Drug Resist. 2020, 13, 2485–2493.
- Kvedaraite, E.; Hertwig, L.; Sinha, I.; Ponzetta, A.; Hed, I.; Lourda, M.; Dzidic, M.; Akber, M.; Klingstrom, J.; Bjorkstrom, N.K.; et al. Major Alterations in the Mononuclear Phagocyte Landscape Associated with COVID-19 Severity. Proc. Natl. Acad. Sci. USA 2021, 118, e2018587118.
- Kim, D.-M.; Kim, Y.; Seo, J.-W.; Lee, J.; Park, U.; Ha, N.Y.; Koh, J.; Park, H.; Lee, J.W.; Ro, H.J.; et al. Enhanced Eosinophil-Mediated Inflammation Associated with Antibody and Complement-Dependent Pneumonic Insults in Critical COVID-19. Cell Rep. 2021, 37, 109798.
- Eguíluz-Gracia, I.; Malmstrom, K.; Dheyauldeen, S.A.; Lohi, J.; Sajantila, A.; Aaløkken, R.; Sundaram, A.Y.M.; Gilfillan, G.D.; Makela, M.; Baekkevold, E.S.; et al. Monocytes Accumulate in the Airways of Children with Fatal Asthma. Clin. Exp. Allergy 2018, 48, 1631–1639.
- Niessen, N.M.; Baines, K.J.; Simpson, J.L.; Scott, H.A.; Qin, L.; Gibson, P.G.; Fricker, M. Neutrophilic Asthma Features Increased Airway Classical Monocytes. Clin. Exp. Allergy 2021, 51, 305–317.
- Zhou, R.; To, K.K.W.; Wong, Y.C.; Liu, L.; Zhou, B.; Li, X.; Huang, H.; Mo, Y.; Luk, T.Y.; Lau, T.T.K.; et al. Acute SARS-CoV-2 Infection Impairs Dendritic Cell and T Cell Responses. Immunity 2020, 53, 864–877.e5.
- Xu, G.; Qi, F.; Li, H.; Yang, Q.; Wang, H.; Wang, X.; Liu, X.; Zhao, J.; Liao, X.; Liu, Y.; et al. The Differential Immune Responses to COVID-19 in Peripheral and Lung Revealed by Single-Cell RNA Sequencing. Cell Discov. 2020, 6, 73.
- Gómez-Rial, J.; Currás-Tuala, M.J.; Rivero-Calle, I.; Gómez-Carballa, A.; Cebey-López, M.; Rodríguez-Tenreiro, C.; Dacosta-Urbieta, A.; Rivero-Velasco, C.; Rodríguez-Núñez, N.; Trastoy-Pena, R.; et al. Increased Serum Levels of SCD14 and SCD163 Indicate a Preponderant Role for Monocytes in COVID-19 Immunopathology. Front. Immunol. 2020, 11, 560381.
- Niles, M.A.; Gogesch, P.; Kronhart, S.; Iannazzo, S.O.; Kochs, G.; Waibler, Z.; Anzaghe, M. Macrophages and Dendritic Cells Are Not the Major Source of Pro-In Fl Ammatory Cytokines Upon SARS-CoV-2 Infection. Front. Immunol. 2021, 12, 647824.
- Winheim, E.; Rinke, L.; Lutz, K.; Reischer, A.; Leutbecher, A.; Wolfram, L.; Rausch, L.; Kranich, J.; Wratil, P.R.; Huber, J.E.; et al. Impaired Function and Delayed Regeneration of Dendritic Cells in COVID-19. PLoS Pathog. 2021, 17, e1009742.
- Knoll, R.; Schultze, J.L.; Schulte-Schrepping, J. Monocytes and Macrophages in COVID-19. Front. Immunol. 2021, 12, 720109.
- Merad, M.; Martin, J.C. Pathological Inflammation in Patients with COVID-19: A Key Role for Monocytes and Macrophages. Nat. Rev. Immunol. 2020, 20, 355–362.
- Tay, M.Z.; Poh, C.M.; Rénia, L.; Macary, P.A.; Ng, L.F.P. The Trinity of COVID-19: Immunity, Inflammation and Intervention. Nat. Rev. Immunol. 2020, 30, 363–374.
- Theobald, S.J.; Simonis, A.; Georgomanolis, T.; Kreer, C.; Zehner, M.; Eisfeld, H.S.; Albert, M.; Chhen, J.; Motameny, S.; Winter, S.; et al. Long-Lived Macrophage Reprogramming Drives Spike Protein-Mediated Inflammasome Activation in COVID-19. EMBO Mol. Med. 2021, 13, e14150.
- Shah, A. Novel Coronavirus-Induced NLRP3 Inflammasome Activation: A Potential Drug Target in the Treatment of COVID-19. Front. Immunol. 2020, 11, 1021.
- Nieto-Torres, J.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Castaño-Rodriguez, C.; Fernandez-Delgado, R.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Severe Acute Respiratory Syndrome Coronavirus E Protein Transports Calcium Ions and Activates the NLRP3 Inflammasome. Virology 2015, 485, 330–339.
- Hoel, H.; Heggelund, L.; Reikvam, D.H.; Stiksrud, B.; Ueland, T.; Michelsen, A.E.; Otterdal, K.; Muller, K.E.; Lind, A.; Muller, F.; et al. Elevated Markers of Gut Leakage and Inflammasome Activation in COVID-19 Patients with Cardiac Involvement. J. Intern. Med. 2021, 289, 523–531.
- Wang, J.; Xu, Y.; Zhang, X.; Wang, S.; Peng, Z.; Guo, J.; Jiang, H.; Liu, J.; Xie, Y.; Wang, J.; et al. Leptin Correlates with Monocytes Activation and Severe Condition in COVID-19 Patients. J. Leukoc. Biol. 2021, 110, 9–20.
- Lv, J.; Wang, Z.; Qu, Y.; Zhu, H.; Zhu, Q.; Tong, W.; Bao, L.; Lv, Q.; Cong, J.; Li, D.; et al. Distinct Uptake, Amplification, and Release of SARS-CoV-2 by M1 and M2 Alveolar Macrophages. Cell Discov. 2021, 7, 24.
- Paula, C.B.V.; Azevedo, M.L.V.; Nagashima, S.; Martins, A.P.C.; Malaquias, M.A.S.; Miggiolaro, A.F.R.D.S.; da Silva Motta Júnior, J.; Avelino, G.; do Carmo, L.A.P.; Carstens, L.B.; et al. IL-4/IL-13 Remodeling Pathway of COVID-19 Lung Injury. Sci. Rep. 2020, 10, 4–11.
- Patente, T.A.; Pinho, M.P.; Oliveira, A.A.; Evangelista, G.C.M.; Bergami-Santos, P.C.; Barbuto, J.A.M. Human Dendritic Cells: Their Heterogeneity and Clinical Application Potential in Cancer Immunotherapy. Front. Immunol. 2019, 9, 3176.
- Borges, R.C.; Hohmann, M.S.; Borghi, S.M. Dendritic Cells in COVID-19 Immunopathogenesis: Insights for a Possible Role in Determining Disease Outcome. Int. Rev. Immunol. 2021, 40, 108–125.
- Marongiu, L.; Protti, G.; Facchini, F.A.; Valache, M.; Mingozzi, F.; Ranzani, V.; Mancini, N.; Abrignani, S.; Spreafico, R.; Granucci, F. Maturation Signatures of Conventional Dendritic Cell Subtypes in COVID-19 Suggest Direct Viral Sensing. Eur. J. Immunol. 2022, 52, 109–122.
- Severa, M.; Diott, R.A.; Etna, M.P.; Rizzo, F.; Fiore, S.; Ricci, D.; Iannetta, M.; Sinigaglia, A.; Lodi, A.; Mancini, N.; et al. Differential Plasmacytoid Dendritic Cell Phenotype and Type I Interferon Response in Asymptomatic and Severe COVID-19 Infection. PLoS Pathog. 2021, 17, e1009878.
- Bitirgen, G.; Korkmaz, C.; Zamani, A.; Ozkagnici, A.; Zengin, N.; Ponirakis, G.; Malik, R.A. Corneal Confocal Microscopy Identifies Corneal Nerve Fibre Loss and Increased Dendritic Cells in Patients with Long COVID. Br. J. Ophthalmol. 2021, 106, 1635–1641.
- Arunachalam, P.S.; Wimmers, F.; Mok, C.K.P.; Perera, R.A.P.M.; Scott, M.; Hagan, T.; Sigal, N.; Feng, Y.; Bristow, L.; Tsang, O.T.Y.; et al. Systems Biological Assessment of Immunity to Mild versus Severe COVID-19 Infection in Humans. Science 2020, 369, 1210–1220.
- Pérez-gómez, A.; Vitallé, J.; Gasca-capote, C.; Gutierrez-valencia, A.; Trujillo-rodriguez, M.; Serna-gallego, A.; Muñoz-muela, E.; Jiménez-leon, M.D.L.R.; Ra, M.; Rivas-jeremias, I.; et al. Dendritic Cell Deficiencies Persist Seven Months after SARS-CoV-2 Infection. Cell. Mol. Immunol. 2021, 18, 2128–2139.
- Jouan, Y.; Guillon, A.; Gonzalez, L.; Perez, Y.; Boisseau, C.; Ehrmann, S.; Ferreira, M.; Daix, T.; Jeannet, R.; François, B.; et al. Phenotypical and Functional Alteration of Unconventional T Cells in Severe COVID-19 Patients. J. Exp. Med. 2020, 217, e20200872.
- Rojas, M.; Rodríguez, Y.; Monsalve, D.M.; Acosta-Ampudia, Y.; Camacho, B.; Gallo, J.E.; Rojas-Villarraga, A.; Ramírez-Santana, C.; Díaz-Coronado, J.C.; Manrique, R.; et al. Convalescent Plasma in COVID-19: Possible Mechanisms of Action. Autoimmun. Rev. 2020, 19, 102554.
More
Information
Subjects:
Immunology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Entry Collection:
COVID-19
Revisions:
2 times
(View History)
Update Date:
29 Mar 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No