Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Cameron Keighron | -- | 1227 | 2023-03-21 16:40:15 | | | |
| 2 | Conner Chen | Meta information modification | 1227 | 2023-03-22 02:59:25 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Keighron, C.N.; Avazzadeh, S.; Goljanek-Whysall, K.; Mcdonagh, B.; Howard, L.; Ritter, T.; Quinlan, L.R. Aetiology and Pathophysiology of Parkinson’s and Alzheimer’s Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/42399 (accessed on 27 July 2026).
Keighron CN, Avazzadeh S, Goljanek-Whysall K, Mcdonagh B, Howard L, Ritter T, et al. Aetiology and Pathophysiology of Parkinson’s and Alzheimer’s Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/42399. Accessed July 27, 2026.
Keighron, Cameron Noah, Sahar Avazzadeh, Katarzyna Goljanek-Whysall, Brian Mcdonagh, Linda Howard, Thomas Ritter, Leo R. Quinlan. "Aetiology and Pathophysiology of Parkinson’s and Alzheimer’s Disease" Encyclopedia, https://encyclopedia.pub/entry/42399 (accessed July 27, 2026).
Keighron, C.N., Avazzadeh, S., Goljanek-Whysall, K., Mcdonagh, B., Howard, L., Ritter, T., & Quinlan, L.R. (2023, March 21). Aetiology and Pathophysiology of Parkinson’s and Alzheimer’s Disease. In Encyclopedia. https://encyclopedia.pub/entry/42399
Keighron, Cameron Noah, et al. "Aetiology and Pathophysiology of Parkinson’s and Alzheimer’s Disease." Encyclopedia. Web. 21 March, 2023.
Copy Citation
Neurodegeneration is hallmarked by the progressive loss of dopaminergic neurons and/or a significant increase in protein aggregates in the brain. Neurodegenerative diseases are a leading cause of death worldwide with over 15 million people currently suffering from either Parkinson’s disease (PD) or Alzheimer’s disease (AD). PD is often characterized by both motor and non-motor symptoms, including muscle rigidity, tremors and bradykinesia, with AD displaying symptoms of confusion and dementia.
Parkinson’s
Alzheimer’s
neurodegeneration
1. Introduction
The hallmark of neurodegeneration is the progressive loss of brain function, often with overlapping biological and clinical symptoms [1]. In general, neurodegenerative diseases share common pathologies and pathways, most prominently protein aggregation, oxidative stress, neuroinflammation and blood–brain barrier dysfunction, leading primarily to the death of neurons from various regions of the brain [2][3][4]. Neuronal degeneration is profound in both Alzheimer’s (AD) and Parkinson’s disease (PD) with numerous overlapping pathways implicated, such as regional aggregation of intracellular proteins Tau and Alpha-synuclein [5]; complex genotype–phenotype relationships with common mutations such as leucine-rich repeat kinase 2 (LRRK2), PTEN-induced kinase 1 (PINK1) and amyloid precursor protein (APP) [6]; and alterations in pathways such as autophagy–lysosome activity, creating an imbalance between autophagosome formation and the autophagic degradation usually involved in clearing aggregated proteins [7]. Mitochondrial homeostasis is also implicated in AD and PD, leading to neuronal cell death via an increase in oxidative stress [8][9]. Lastly, innate immunity, synaptic toxicity and network dysfunction all contribute to the neuronal loss observed in AD and PD [10] (Figure 1).
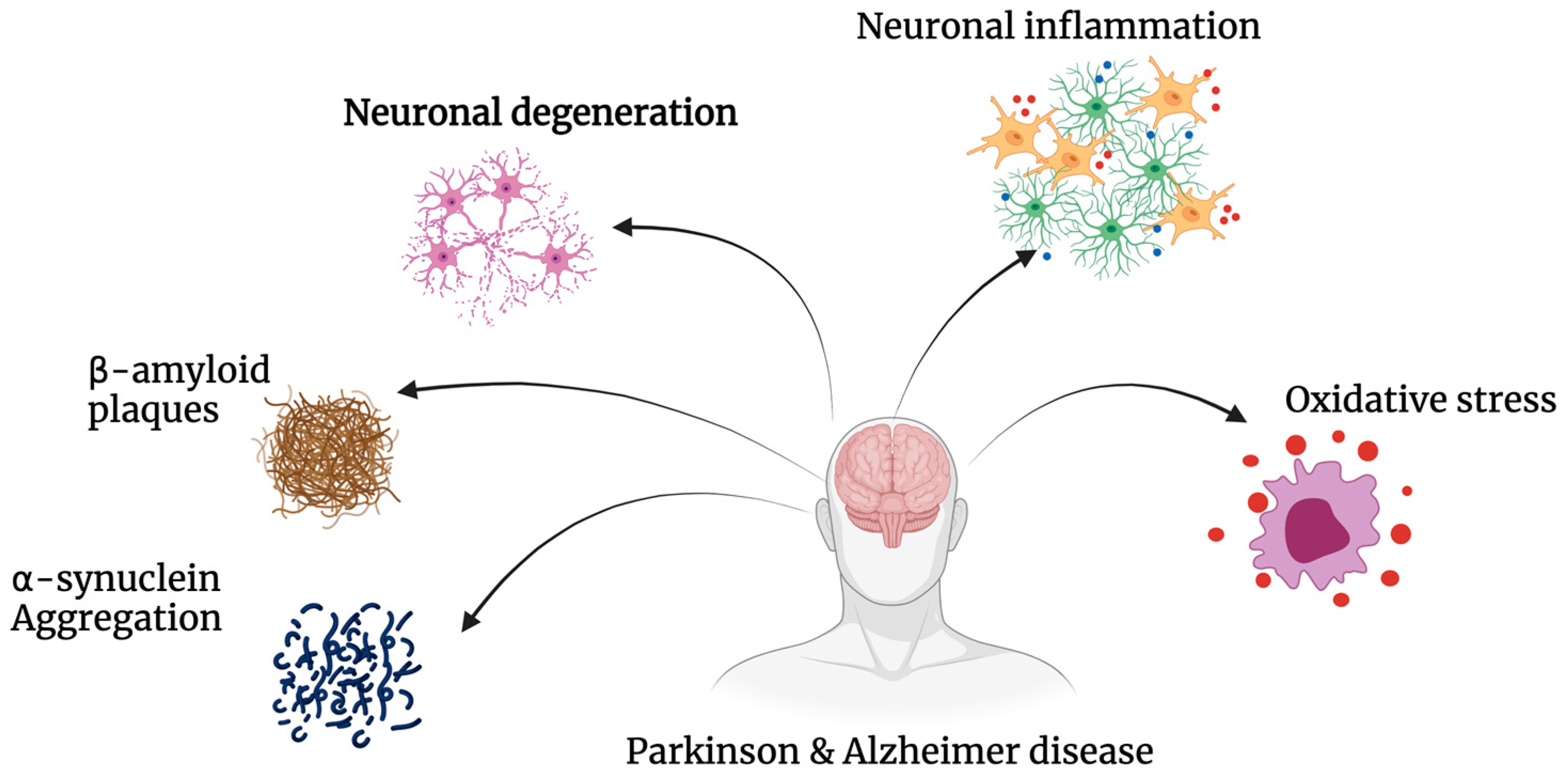
Figure 1. Pathogenesis of Alzheimer’s’ disease and Parkinson’s’ disease. AD and PD share common pathways of degeneration. Both neurodegenerative diseases exhibit significant amounts of oxidative stress, neuronal inflammation and degeneration, as well as the build-up of insoluble proteins including β-amyloid and α-synuclein.
AD and PD are the two most common neurodegenerative diseases worldwide. Cognitive dysfunction is the primary symptom exhibited in AD, while significant motor dysfunction is cardinal to PD [5]. AD is the most common cause of dementia, with a prevalence estimated at 24 million worldwide, which is expected to continue to rise over the next decade (GBD 2019 Dementia Forecasting Collaborators 2022). AD affects around 11% of the population over the age of 65, with PD affecting 2–3% of the population over 65 years of age, becoming the second most common neurodegenerative disease after AD [11]. AD can be classified into four stages: pre-clinical/pre-symptomatic, mild/early stage, moderate stage and severe/late stage [12][13][14]. These stages are often characterised by progressive memory loss, impaired balance, aphasia-like symptoms and an overall lack of independence in carrying out activities of daily living [15][16][17]. Two neuropathological changes have been identified in AD; positive lesions and negative lesions. Accumulation of neurofibrillary tangles, amyloid plaques and other deposits are significant indicators of AD, while on the other hand, neuronal, neuropil and synaptic loss-induced atrophy is also indicated in AD [18][19]. While the precise cause of the underlying pathological changes in AD is unknown, risk factors including age, genetics, traumatic brain injuries, diet and immune system dysregulation are some of the key contributors [20].
PD is associated with a lack of dopamine and an overall slowing of movement (bradykinesia) along with either a resting tremor or rigidity [21]. It has been suggested that there are two stages to PD, early (1 and 2) and late (3 and 4). In the early stages, symptoms can include rapid eye movement sleep behaviour disease (including sleep paralysis), as well as decreased smell, suggesting onset in the medulla and olfactory bulb. In stages 3 and 4, symptoms are more typical of cognitive impairment, including issues with movement and gait as well as hallucinations, suggesting that this stage’s pathology has progressed to the substantia nigra pars compacta and other midbrain and basal forebrain regions [22][23], often with aggregates of alpha-synuclein [23].
2. Aetiology and Pathophysiology of Disease
PD and AD are characterised by the presence of insoluble protein deposits, β-amyloid plaques and tau-containing neurofibrillary lesions in AD and α-synuclein-rich Lewy bodies in PD (Figure 1). Neuropathological changes in the disease progression and pathology of AD include neurofibrillary tangles, amyloid plaques, dystrophic neurites and neuropil threads [24]. PD presents with abnormal α-synuclein aggregates and the presence of Lewy bodies with selective loss of dopaminergic neurons [25][26]. Typically, PD is diagnosed at the mid to late stage of the disease due to a long period of dormancy between the initial loss of dopaminergic neurons and the development of more “typical” clinical symptoms [21][27]. AD follows a similar pathway with initial symptoms presenting over a 2–4-year period, then progressively worsening over the subsequent 10 years. PD and AD are largely considered to be sporadic, with evidence in recent years continuing to support the hypothesis that AD and PD may have substantial genetic components. In PD, this includes reported defects in the SNCA, PINK1 genes (associated with abnormal mitochondria and increased apoptosis), parkin (associated with impaired damaged protein tagging with ubiquitin), LRRK2 (associated with increased neuroinflammation) and DJ-1 (associated with increased reactive oxygen species), with genes such as APP (associated with the generation of beta-amyloid peptides), PSEN1 and PSEN2 (both interact with APP and are associated with the overproduction of toxic beta-amyloid peptides) displaying defects in AD [28][29][30][31].
Taking a closer look at both AD and PD suggests many commonalities in their pathophysiology, with many genes shared and co-expressed [32], as well as stark differences [33]. AD is well known for its increased concentration of Aβ42 (β-amyloid 42—due to mutations in the APP gene) which encourages the production of oligomers (that are neurotoxic). These oligomers cluster and eventually form plaques that contribute to symptoms presented above for AD [34][35]. Tau, usually functioning to stabilise axonal microtubules, is abnormally phosphorylated in AD. When this occurs, these highly phosphorylated tau proteins tend to clump together into filaments and further into insoluble neurofibrillary aggregates, spreading throughout the brain [36][37]. Tauopathy is also closely linked to granulo-vacuolar degeneration (GVD) in AD patients. Granulo-vacuolar bodies (GVBs) are often present in hippocampal pyramidal cells of those with AD, which is likely associated with cognitive decline in patients [34][38][39].
The increased loss of neurons in the substania nigra pars compacta (mostly), combined with the presence of abnormal aggregates, underpins the pathophysiology of PD [40]. Losing these neurons, predominately dopaminergic neurons, reduces dopamine levels, thus leading to the symptoms [41]. Drilling down into mechanisms, alterations in α-synuclein lead to either an increase in aggregation of protein or diminished capacity for its degradation, creating fibrilization of α-synuclein in Lewy bodies or neurites promoting neurodegeneration [42][43]. Furthermore, this α-synuclein accumulation directly increases levels of mitochondrial stress and reactive oxygen species [44][45]. Parkin is associated with many subcellular compartments such as synaptic vesicles and the endoplasmic reticulum, as well as playing a neuroprotective role via mitochondria by delaying mitochondrial swelling and the activation of caspase 3 [46]. Its function is closely linked to the ubiquitin-proteasome proteolytic pathway. Parkin is an E3 ubiquitin-protein ligase that, in partnership with E2-conjugating enzymes, selects protein substrates for ubiquitylation and their subsequent degradation [46][47][48]. Oxidative stress transforms DJ-1 (which has a key role in antioxidant activities as well as in directly inhibiting α-synuclein aggregation) into a more acidic isoform which translocates to the outer mitochondrial membrane with PINK1 (located on the inner membrane of mitochondria), regulating the function of mitochondria through phosphorylation of substrates. In neurodegeneration associated with PD, one or more of these pathways is impaired; losing parkin results in the accumulation of mitochondrial substrates, exposing mitochondria to increased stress, and proteasomal impairment amplifies this effect. A reduction in ATP exacerbates the proteasomal impairment, which leads to an increase in α-synuclein accumulation [43][49][50][51][52][53] and a further increase in neurodegeneration.
References
- Dugger, B.N.; Dickson, D.W. Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035.
- Kurosinski, P.; Guggisberg, M.; Götz, J. Alzheimer’s and Parkinson’s disease—Overlapping or synergistic pathologies? Trends Mol. Med. 2002, 8, 3–5.
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative Stress and Neurodegenerative Diseases: A Review of Upstream and Downstream Antioxidant Therapeutic Options. Curr. Neuropharmacol. 2009, 7, 65–74.
- Tan, S.H.; Karri, V.; Tay, N.W.R.; Chang, K.H.; Ah, H.Y.; Ng, P.Q.; Ho, H.S.; Keh, H.W.; Candasamy, M. Emerging pathways to neurodegeneration: Dissecting the critical molecular mechanisms in Alzheimer’s disease, Parkinson’s disease. Biomed. Pharmacother. 2019, 111, 765–777.
- Xie, A.; Gao, J.; Xu, L.; Meng, D. Shared mechanisms of neurodegeneration in Alzheimer’s disease and Parkinson’s disease. Biomed. Res. Int. 2014, 2014, 648740.
- Guadagnolo, D.; Piane, M.; Torrisi, M.R.; Pizzuti, A.; Petrucci, S. Genotype-Phenotype Correlations in Monogenic Parkinson Disease: A Review on Clinical and Molecular Findings. Front. Neurol. 2021, 12, 648588.
- Kiriyama, Y.; Nochi, H. The Function of Autophagy in Neurodegenerative Diseases. Int. J. Mol. Sci. 2015, 16, 26797–26812.
- Scorziello, A.; Borzacchiello, D.; Sisalli, M.J.; Di Martino, R.; Morelli, M.; Feliciello, A. Mitochondrial Homeostasis and Signaling in Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 100.
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Parkinsons Dis. 2013, 3, 461–491.
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309.
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013.
- Apostolova, L.G. Alzheimer Disease. Continuum 2016, 22, 419–434.
- Wattmo, C.; Minthon, L.; Wallin, K. Mild versus moderate stages of Alzheimer’s disease: Three-year outcomes in a routine clinical setting of cholinesterase inhibitor therapy. Alzheimer Res. Ther. 2016, 8, 7.
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016, 12, 292–323.
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944.
- Neugroschl, J.; Wang, S. Alzheimer’s disease: Diagnosis and treatment across the spectrum of disease severity. Mt. Sinai J. Med. 2011, 78, 596–612.
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789.
- Singh, S.K.; Srivastav, S.; Yadav, A.K.; Srikrishna, S.; Perry, G. Overview of alzheimer’s disease and some therapeutic approaches targeting aβ by using several synthetic and herbal compounds. Oxid. Med. Cell. Longev. 2016, 2016, 7361613.
- Spires-Jones, T.L.; Hyman, B.T. The Intersection of Amyloid Beta and Tau at Synapses in Alzheimer’s Disease. Neuron 2014, 82, 756–771.
- Armstrong, R.A. Risk factors for Alzheimer’s disease. Folia Neuropathol. 2019, 57, 87–105.
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s disease: Biomarkers, treatment, and risk factors. Front. Neurosci. 2018, 12, 612.
- Armstrong, M.J.; Okun, M.S. Diagnosis and treatment of parkinson disease: A review. JAMA 2020, 323, 548–560.
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211.
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189.
- Stoker, T.B.; Torsney, K.M.; Barker, R.A. Emerging Treatment Approaches for Parkinson’s Disease. Front. Neurosci. 2018, 12, 693.
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840.
- El-Agnaf, O.M.A.; Salem, S.A.; Paleologou, K.E.; Curran, M.D.; Gibson, M.J.; Court, J.A.; Schlossmacher, M.G.; Allsop, D. Detection of oligomeric forms of α-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J. 2006, 20, 419–425.
- Bekris, L.M.; Mata, I.F.; Zabetian, C.P. The genetics of Parkinson disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 228–242.
- Simón-Sánchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312.
- Piaceri, I.; Nacmias, B.; Sorbi, S. Genetics of familial and sporadic Alzheimer s disease. Front. Biosci. 2013, E5, 167–177.
- Avazzadeh, S.; Baena, J.M.; Keighron, C.; Feller-Sanchez, Y.; Quinlan, L.R. Modelling Parkinson’s Disease: iPSCs towards Better Understanding of Human Pathology. Brain Sci. 2021, 11, 373.
- Morgan, S.L.; Naderi, P.; Koler, K.; Pita-Juarez, Y.; Prokopenko, D.; Vlachos, I.S.; Tanzi, R.E.; Bertram, L.; Hide, W.A. Most pathways can be related to the pathogenesis of alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 846902.
- Ahmad, K.; Baig, M.H.; Mushtaq, G.; Kamal, M.A.; Greig, N.H.; Choi, I. Commonalities in Biological Pathways, Genetics, and Cellular Mechanism between Alzheimer Disease and Other Neurodegenerative Diseases: An In Silico-Updated Overview. Curr. Alzheimer Res. 2017, 14, 1190–1197.
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185.
- De Strooper, B.; Vassar, R.; Golde, T. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99–107.
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404.
- Alonso, A.D.C.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 1996, 2, 783–787.
- Wiersma, V.I.; Hoozemans, J.J.M.; Scheper, W. Untangling the origin and function of granulovacuolar degeneration bodies in neurodegenerative proteinopathies. Acta Neuropathol. Commun. 2020, 8, 153.
- Castellani, R.J.; Gupta, Y.; Sheng, B.; Siedlak, S.L.; Harris, P.L.; Coller, J.M.; Perry, G.; Lee, H.-G.; Tabaton, M.; Smith, M.A.; et al. A novel origin for granulovacuolar degeneration in aging and Alzheimer’s disease: Parallels to stress granules. Lab. Investig. 2011, 91, 1777–1786.
- Lang, A.E.; Lozano, A.M. Parkinson’s disease. First of two parts. N. Engl. J. Med. 1998, 339, 1044–1053.
- Hoang, Q.Q. Pathway for Parkinson disease. Proc. Natl. Acad. Sci. USA 2014, 111, 2402–2403.
- Shastry, B.S. Parkinson disease: Etiology, pathogenesis and future of gene therapy. Neurosci. Res. 2001, 41, 5–12.
- Corti, O.; Hampe, C.; Darios, F.; Ibanez, P.; Ruberg, M.; Brice, A. Parkinson’s disease: From causes to mechanisms. Comptes Rendus Biol. 2005, 328, 131–142.
- Park, J.-H.; Burgess, J.D.; Faroqi, A.H.; DeMeo, N.N.; Fiesel, F.C.; Springer, W.; Delenclos, M.; McLean, P.J. Alpha-synuclein-induced mitochondrial dysfunction is mediated via a sirtuin 3-dependent pathway. Mol. Neurodegener. 2020, 15, 5.
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial Import and Accumulation of alpha-Synuclein Impair Complex I in Human Dopaminergic Neuronal Cultures and Parkinson Disease Brain. J. Biol. Chem. 2008, 283, 9089–9100.
- Darios, F.; Corti, O.; Hampe, C.; Muriel, M.-P.; Abbas, N.; Gu, W.-J.; Lücking, C.B.; Hirsch, E.C.; Rooney, T.; Ruberg, M.; et al. Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum. Mol. Genet. 2003, 12, 517–526.
- Zhang, Y.; Gao, J.; Chung, K.K.K.; Huang, H.; Dawson, V.L.; Dawson, T.M. Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc. Natl. Acad. Sci. USA 2000, 97, 13354–13359.
- Grimaldo, L.; Sandoval, A.; Garza-López, E.; Felix, R. Involvement of Parkin in the ubiquitin proteasome system-mediated degradation of N-type voltage-gated Ca2+ channels. PLoS ONE 2017, 12, e0185289.
- Healy, D.G.; Abou-Sleiman, P.M.; Valente, E.M.; Gilks, W.P.; Bhatia, K.; Quinn, N.; Lees, A.J.; Wood, N.W. DJ-1 mutations in Parkinson’s disease. J. Med. Genet. 2004, 41, 248.
- Höglinger, G.U.; Carrard, G.; Michel, P.P.; Medja, F.; Lombès, A.; Ruberg, M.; Friguet, B.; Hirsch, E. Dysfunction of mitochondrial complex I and the proteasome: Interactions between two biochemical deficits in a cellular model of Parkinson’s disease. J. Neurochem. 2003, 86, 1297–1307.
- McNaught, K.S.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194.
- Abou-Sleiman, P.M.; Healy, D.G.; Quinn, N.; Lees, A.J.; Wood, N.W. The role of pathogenicDJ-1 mutations in Parkinson’s disease. Ann. Neurol. 2003, 54, 283–286.
- Yang, J.; Kim, K.S.; Iyirhiaro, G.O.; Marcogliese, P.C.; Callaghan, S.M.; Qu, D.; Kim, W.J.; Slack, R.S.; Park, D.S. DJ-1 modulates the unfolded protein response and cell death via upregulation of ATF4 following ER stress. Cell Death Dis. 2019, 10, 135.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.9K
Revisions:
2 times
(View History)
Update Date:
22 Mar 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No