Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Manoj K. Pandey | -- | 3283 | 2023-03-16 19:06:26 | | | |
| 2 | Camila Xu | Meta information modification | 3283 | 2023-03-17 03:32:00 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Elbezanti, W.O.; Challagundla, K.B.; Jonnalagadda, S.C.; Budak-Alpdogan, T.; Pandey, M.K. Multiple Myeloma Pathogenesis and The Existing Therapies. Encyclopedia. Available online: https://encyclopedia.pub/entry/42279 (accessed on 09 August 2026).
Elbezanti WO, Challagundla KB, Jonnalagadda SC, Budak-Alpdogan T, Pandey MK. Multiple Myeloma Pathogenesis and The Existing Therapies. Encyclopedia. Available at: https://encyclopedia.pub/entry/42279. Accessed August 09, 2026.
Elbezanti, Weam Othman, Kishore B. Challagundla, Subash C. Jonnalagadda, Tulin Budak-Alpdogan, Manoj K. Pandey. "Multiple Myeloma Pathogenesis and The Existing Therapies" Encyclopedia, https://encyclopedia.pub/entry/42279 (accessed August 09, 2026).
Elbezanti, W.O., Challagundla, K.B., Jonnalagadda, S.C., Budak-Alpdogan, T., & Pandey, M.K. (2023, March 16). Multiple Myeloma Pathogenesis and The Existing Therapies. In Encyclopedia. https://encyclopedia.pub/entry/42279
Elbezanti, Weam Othman, et al. "Multiple Myeloma Pathogenesis and The Existing Therapies." Encyclopedia. Web. 16 March, 2023.
Copy Citation
Multiple myeloma (MM) is a mature B-cell neoplasm that is characterized by uncontrolled growth of plasma cells (PCs) in bone marrow (BM) which leads to excessive secretion of antibodies. The progression of MM is a multistep process that starts with an asymptomatic premalignant condition known as monoclonal gammopathy of undetermined significance (MGUS), in which BM produces abnormal PCs and secretes M protein instead of normal antibodies.
multiple myeloma
B-cell malignancy
bone marrow microenvironment
1. The Pathogenesis of Multiple Myeloma
Multiple myeloma (MM) is a mature B-cell neoplasm that is characterized by uncontrolled growth of plasma cells (PCs) in bone marrow (BM) which leads to excessive secretion of antibodies. The progression of MM is a multistep process that starts with an asymptomatic premalignant condition known as monoclonal gammopathy of undetermined significance (MGUS), in which BM produces abnormal PCs and secretes M protein instead of normal antibodies [1]. With the increase in oncogenic mutations, MGUS evolves into smoldering MM (SMM), which is characterized by a higher serum level of M protein and a higher percentage of clonal PCs. About 50% of patients with SMM show a constant increase of M protein and develop MM [2]. While both MGUS and SMM are asymptomatic, complications of accumulated proteins may start to affect the kidneys [3]. Almost 20–40% of MM patients have renal disease by the time of diagnosis [4].
The complexity of MM is attributed to the clinical and biological heterogeneity of the disease that further genetically evolves during its progression [5]. MM cells have a wide range of genetic changes including point mutations, insertions, deletions, multiploidy, and chromosomal translocations [6]. For example, trisomic MM and patients with t(11;14) are considered standard-risk patients. On the other hand, MM patients with t(4;14), t(14;16), t(14;20), p53 mutation, gain 1q, or del(17p) are considered to be high-risk [7][8]. Moreover, the bone marrow microenvironment (BMM) plays an important role in disease development, progression, and resistance [9]. All these factors enhance different signaling pathways that contribute to proliferation, survival, invasion, angiogenesis, and osteoclastogenesis [10]. There are many signaling pathways that protect against apoptosis and support MM growth which become activated through the adhesion of MM to the BMM. These activated pathways include phosphatidylinositol-3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR), and nuclear factor kappa B (NF-κB), janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3), which support MM growth and protect against apoptosis (Figure 1). The activation of these pathways leads to upregulation and secretion of several cytokines and factors from both MM and BMM cells such as interleukin-6 (IL-6), insulin-like growth factor-1, VEGF, tumor necrosis factor alpha (TNF-α), and transforming growth factor-β [11][12].
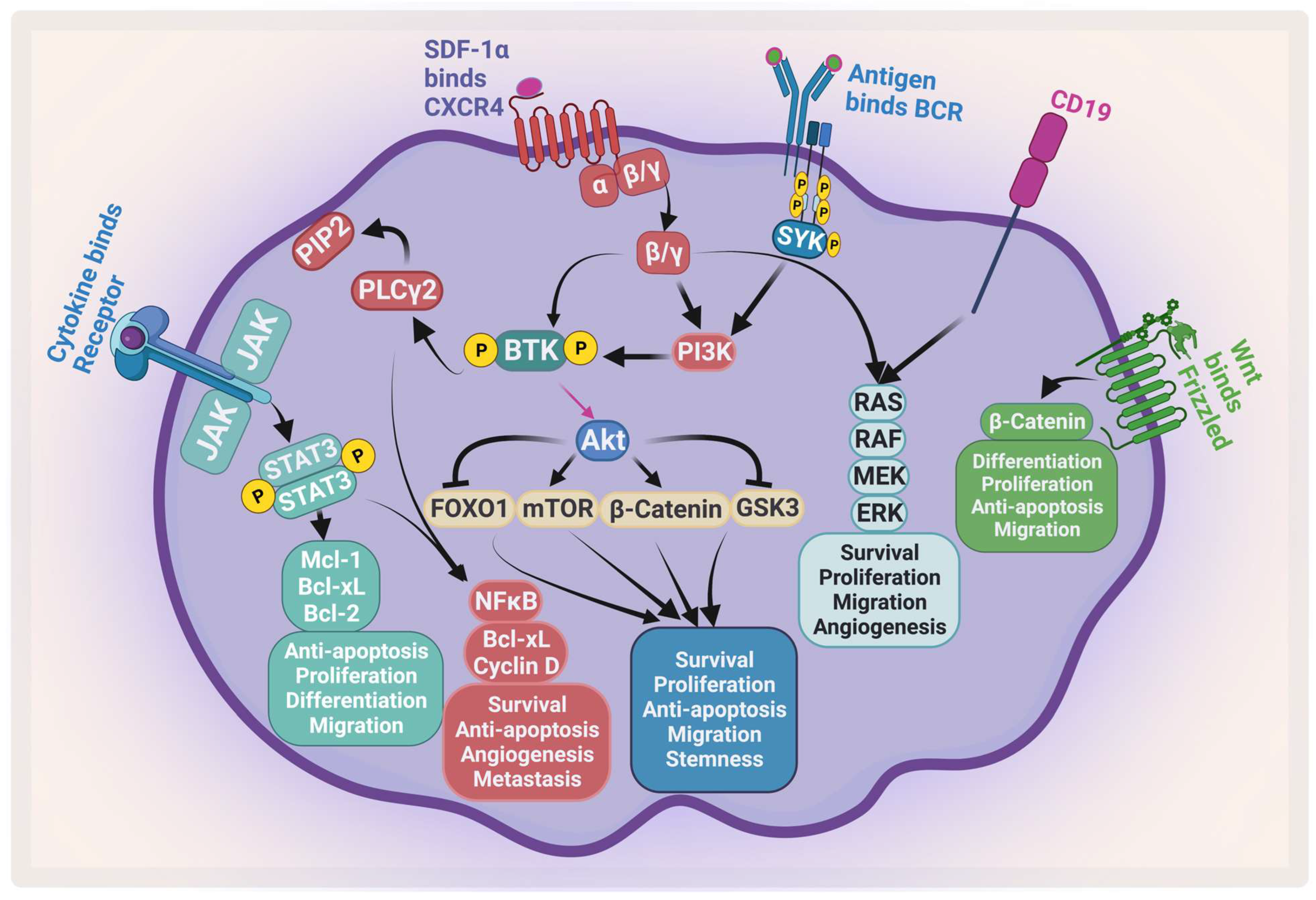
Figure 1. Major signaling pathways in MM. MM receives several survival and proliferation signals: JAK2/STAT3 pathway activates antiapoptotic proteins and activates NF-κB, the PI3K/AKT/mTOR pathways become activated when stromal-derived factor 1 α (SDF-1α) binds to CXCR4 and/or antigen binds to B-cell receptor (BCR). The RAS/MEK/ERK pathway becomes activated by BCR and CD19. Wnt/β-catenin pathway activation enhances differentiation, survival, migration, and antiapoptotic signals. Ras: rat sarcoma; Raf: rapidly accelerated fibrosarcoma; MEK: mitogen-activated protein kinase kinase. Created by Biorender.com.
The BMM contains several specialized cells that are responsible for skeletal integrity, immunity, and blood formation [13][14] (Figure 2). Its compartments are classified into niches: the immune niche, the vascular niche and the endosteal niche [15]. Each niche contains various cell types such as B-cells, T-cells, myeloid-derived suppressor cells, osteoclasts, natural killer (NK) cells, mesenchymal stem cells, BM stromal cells (BMSCs), osteoblasts, and endothelial progenitor cells [16].
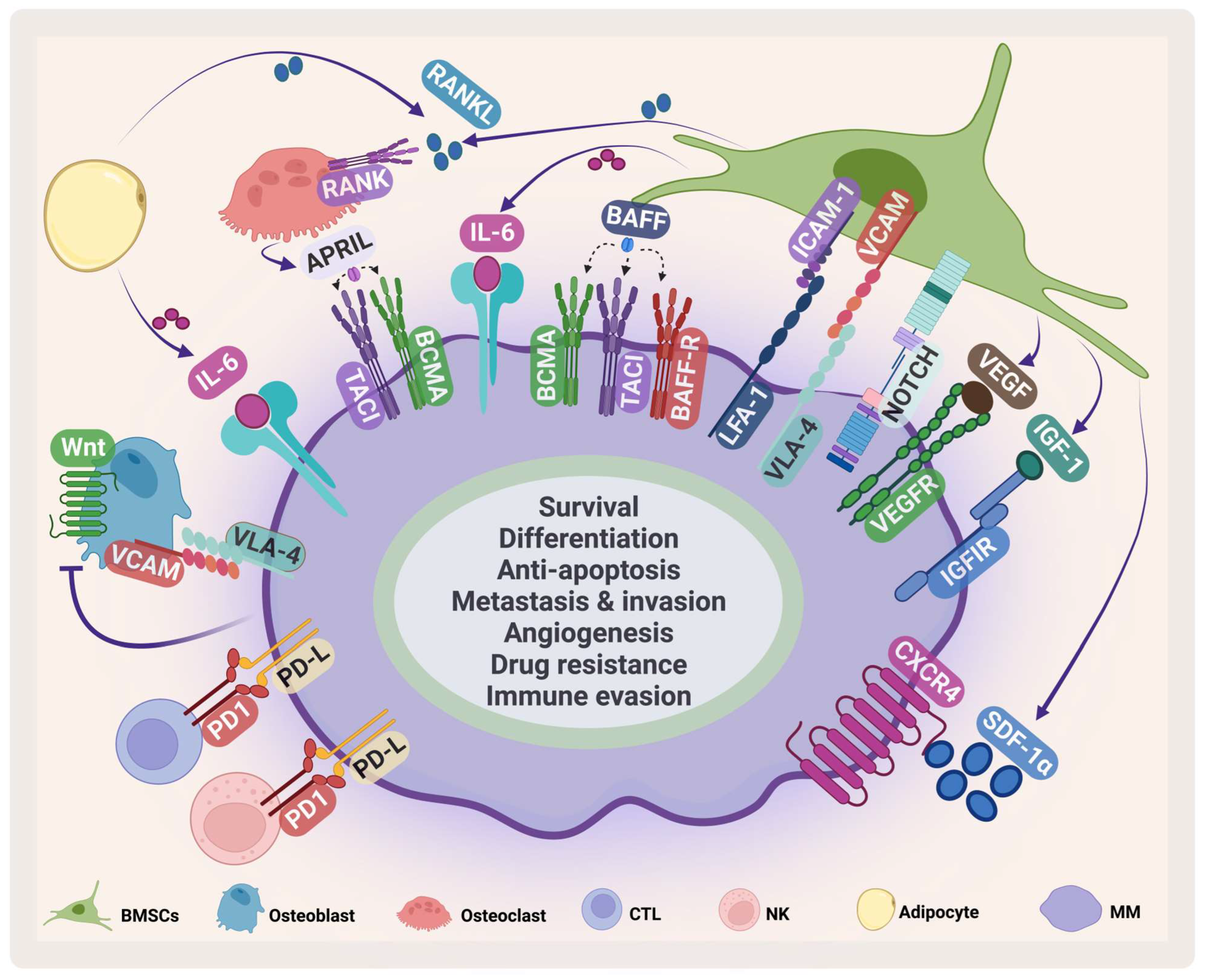
Figure 2. MM microenvironment. MM cells interact with several cells in the BMM such as BMSCs, osteoclasts, osteoblasts, cytotoxic T-cell (CTLs), natural killer (NK) cells, adipocytes. This interaction enhances MM survival and inhibits immune cells. VCAM: vascular cell adhesion protein; LFA-1: lymphocyte function-associated antigen-1; ICAM-1: intercellular adhesion molecule 1; IGF-1: insulin-like growth factor-1. Created by Biorender.com.
BMSCs are a heterogeneous cell population that supports hematopoiesis in normal conditions. They play an important role in supporting the survival, proliferation, and drug resistance of MM [17]. They communicate with MM in several ways. The direct cell adhesion contact through adhesion molecules such as very late antigen 4 (VLA-4), vascular cell adhesion protein, lymphocyte function-associated antigen-1, and intercellular adhesion molecule 1 stimulates IL-6 secretion by BMSCs [18] and mediates drug resistance through cellular-adhesion-mediated drug resistance (CAM-DR) [19]. Stromal cell-derived factor-1α (SDF-1α), a chemokine produced by BMSCs, plays an important role in embryogenesis, angiogenesis, hematopoiesis, and inflammation [20][21]. It stimulates homing and migration of cells through G protein-coupled receptor C-X-C chemokine receptor type 4 (CXCR4) [22]. The SDF-1α/CXCR4 axis plays an important role in survival, angiogenesis, metastasis, invasion, and adhesion in MM (Figure 2) [23]. It has been shown that the SDF-1α level in MM is elevated and this elevation contributes to activating several signaling pathways and induces mitogen-activated protein kinase kinase1/2, AKT phosphorylation, mitogen-activated protein kinase (MAPK), and NF-κB in MM cell lines and patient samples [24][25]. The SDF-1α/CXCR4 axis mediates drug resistance through different pathways that are involved in CAM-DR, affecting adhesion molecules, enhancing IL-6 mediated drug resistance, and stimulating pathways including MAP/extracellular signal-regulated kinase (ERK), wingless/integrated3 (Wnt3)/Ras homolog family member A/Ras homologous -associated protein kinase, and Ras homologous/Ras homologous -kinase [26][27][28][29][30].
Other cytokines and growth factors secreted from BMSCs suppress the immune response and facilitate MM immune evasion. One of the most important cytokines in MM is IL-6, which is secreted by different BM cells including BMSCs, osteoclasts, and macrophages [31]. IL-6 secretion stimulates the JAK/STAT3 pathway, which leads to increased survival and proliferation through the upregulation of Mcl-1, Bcl-xL, Bcl-2, c-Myc, and cyclin D1 [32][33][34][35]. As MM progresses, osteoclast activity increases, which in turn causes bone lesions. During disease progression, an imbalance occurs in receptor activator of NF-κB ligand (RANKL), and osteoprotegerin [36].
In autologous hematopoietic progenitor cell (HPC) transplantation, plerixafor, a specific antagonist of SDF-1α binding to CXCR4, was approved in 2008 to induce hematopoietic stem cells (HSCs) and progenitor cells (HPCs) trafficking. It has been shown that it augments granulocyte colony-stimulating factor (G-CSF)-induced mobilization of HSCs and HPCs [37][38].
2. The Existing Therapies for MM
In the 1960s, oral melphalan, an alkylating agent, in combination with prednisone was considered the frontline treatment for MM [39][40]. Then, FDA-approved thalidomide, an immunomodulatory agent (IMiDs), was introduced in MM therapy. Thalidomide enhanced the overall survival (OS) and showed longer progression-free survival (PFS) regardless of patient age when used in combination with melphalan and prednisolone (clinical trial # NCT00232934, and ISRCTN90692740) [41][42]. Additionally in the 1980s, autologous stem cell transplantation (ASCT) followed by a high dose of therapy was introduced and became the standard of care among younger patients with normal renal function [43][44].
The discovery and the introduction of proteasome inhibitors (PIs) in 2003 has tremendously improved PFS in patients [45]. Bortezomib became the first line of treatment for MM in newly diagnosed MM (NDMM) patients. For relapsed and refractory MM (RRMM) patients, it was used in combination with melphalan and prednisone (Table 1) [46]. After the success of bortezomib, other PIs such as carfilzomib and ixazomib were approved for the MM treatment. In the TOURMALINE-MM1 trial (NCT01564537), oral ixazomib was tested in combination with lenalidomide and dexamethasone (IRd) on RRMM patients and it has significantly improved the PFS (20.6 months in the IRd group vs. 14.7 months in the Rd group (lenalidomide and dexamethasone) at a median follow-up of 14.7 months). The overall response rate (ORR) was 78% in the IRd group and 72% in the Rd group. The median OS was not reached at a median follow-up of approximately 23 months. The additional adverse effects between the two groups were limited and there was a similar quality of life between the IRd and the Rd groups [47].
Table 1. FDA-approved medications for MM from 2006 to 2013.
| Drug | Year | Treatment | Adverse Effects | Refs. |
|---|---|---|---|---|
| Thalidomide | 2006 | NDMM | somnolence, constipation, neuropathy, VTE, and rash | [48][49] |
| Lenalidomide | 2006 | Received one prior therapy | neutropenia, thrombocytopenia, leukopenia, lymphopenia, febrile neutropenia, deep vein thrombosis, pulmonary embolism, atrial fibrillation, constipation, diarrhea, fatigue, pneumonia, hypokalemia, hypocalcemia, muscle weakness, neuropathy, and depression | [50] |
| Doxorubicin | 2007 | RRMM | thrombocytopenia, neutropenia, anemia, fatigue, pyrexia, nausea, vomiting, mucositis/stomatitis diarrhea, and hand foot syndrome | [49][51] |
| Bortezomib | 2008 | NDMM | asthenic conditions, diarrhea, constipation, PSN, vomiting, nausea psychiatric disorders, pyrexia, anorexia, thrombocytopenia, leukopenia, neuralgia, neutropenia, and anemia | [49][52] |
| Plerixafor | 2008 | MM | diarrhea, vomiting, nausea, fatigue, headache, injection site reactions, dizziness, and arthralgia | [49][53] |
| Carfilzomib | 2012 | RRMM | fatigue, anemia, nausea, thrombocytopenia, dyspnea, diarrhea, pyrexia, pneumonia, ARF, pyrexia, and CHF | [54] |
| Pomalidomide | 2013 | RRMM | asthenia, fatigue, neutropenia, anemia, constipation, diarrhea, nausea, URTI, dyspnea, back pain, and pyrexia | [55] |
NDMM: newly diagnosed multiple myeloma; RRMM: relapsed/refractory multiple myeloma; NTE: non-transplant-eligible; TE: transplant eligible; Td: thalidomide, dexamethasone; Rd: lenalidomide, dexamethasone; V: bortezomib; PLD-V: pegylated liposomal doxorubicin, bortezomib; MP: melphalan, prednisone; VMP: bortezomib, melphalan, prednisone; G-CSF: granulocyte-colony-stimulating factor; Plerix-G-CSF: plerixafor, granulocyte-colony-stimulating factor; Pd: pomalidomide, dexamethasone; VTE: venous thromboembolism; PSN: Peripheral sensory neuropathy; ARF: acute renal failure; CHF: congestive heart failure; URTI: Upper respiratory tract infection.
The introduction of a new generation of IMiDs such as lenalidomide in 2005, in combination with PIs, increased survival from 14.8 to 30.9 months [56]. Currently, a triple therapy (PI, IMiD, and corticosteroids) is the first line of treatment for MM followed by autologous stem cell transplant (ASCT). Lenalidomide is usually recommended as a maintenance therapy for MM patients [57]. A combination of bortezomib, lenalidomide, and dexamethasone (VRd) was tested on NDMM in the SWOG-S077 phase III clinical trial (NCT00644228) versus Rd. There was a significant improvement in median PFS (43 months in VRd group vs. 30 months in Rd group) and in median OS (75 months in VRd vs. 64 months in the control group). The ORR was 82% in the VRd group vs. 72% in the Rd group [58]. The next generation IMiD, pomalidomide is shown to be effective and is one of the treatment options that is usually considered in combination after the first relapse for patients who are refractory to lenalidomide [59]. It was first approved in 2013 in combination with dexamethasone for RRMM patients [60]. Then, it was approved in combination with anti-CD38 monoclonal antibody and a steroid for RRMM patients who have previously received two therapies including lenalidomide and bortezomib [60][61][62][63].
Introducing daratumumab, an anti-CD38, in clinical trials (MAIA, ALCYONE, CASTOR, and POLLUX) with different combinations improved minimal residual disease negativity (MRD), and PFS [64] (Table 2). In 2019, daratumumab, lenalidomide, and dexamethasone (DRd) treatment was approved in NDMM patients who are ineligible for transplant after phase III MAIA trial (NCT02252172). In this study, DRd showed significant improvement in PFS (not reached) compared with lenalidomide and dexamethasone (Rd) (31.9 months). The median OS was not reached at a median follow-up of 56.2 months. The common adverse effects of this treatment are neutropenia, pneumonia, anemia, and lymphopenia. Treatment-related-death was 4% in the DRd group compared to 3% in the Rd control group [65][66]. In addition, daratumumab was tested in combination with bortezomib and melphalan-prednisone (D-VMP) in a phase III ALCYONE trial (NCT02195479) in NDMM patients. The 18-month PFS was 71.6%. At a median follow-up of 16.5 months, 22.3% of the patients were negative for MRD. The common adverse effects were neutropenia, thrombocytopenia, and anemia [67]. After the CASSIOPEIA phase III trial (NCT02541383) on ND transplant-eligible MM patients, daratumumab was approved to be used in combination with bortezomib, thalidomide, and dexamethasone (D-VTd) in 2019. At a median follow-up of 35.4 months, PFS was not reached versus 46.7 months with the control group. The most common adverse effects were lymphopenia, hypertension, and neutropenia [68].
Table 2. FDA-approved medications for MM from 2014 to 2019.
| Drug | Year | Treatment | Adverse Effects | Name and NCT Number |
Refs. |
|---|---|---|---|---|---|
| Panobinostat | 2015 | RRMM | pneumonia, diarrhea, arrhythmias, hypophosphatemia and hypokalemia, ECG change, thrombocytopenia, neutropenia fatigue, and sepsis | PANORAMA1 NCT01023308 |
[69] |
| Carfilzomib | 2015 | RRMM | CVE, VTE, ARF, pulmonary toxicities, hypertension, and thrombocytopenia | ASPIRE NCT01080391 |
[70] |
| Daratumumab | 2015 | RRMM | fatigue, nausea, back pain, pyrexia, URTI, cough, IRs, lymphopenia, neutropenia, anemia, and thrombocytopenia | SIRIUS NCT01985126 |
[71] |
| Ixazomib | 2015 | RRMM | diarrhea, constipation, thrombocytopenia, PSN, nausea, peripheral edema, vomiting, and back pain | TOURMALITOURMALINE NCT01564537 |
[47] |
| Elotuzumab | 2015 | RRMM | ARF, pneumonia, nasopharyngitis pyrexia, anemia, pulmonary embolism, and PSN | ELOQUENT-2 NCT01239797 |
[72] [73] |
| Daratumumab | 2016 | RRMM | URTI, cough, diarrhea, fatigue, nausea, pyrexia, muscle spasm, and dyspnea, neutropenia, anemia | POLLUX NCT02076009 |
[74] |
| Daratumumab | 2016 | RRMM | URTI, IRs, diarrhea, peripheral edema, Neutropenia, and thrombocytopenia, anemia | CASTOR NCT02136134 |
[75][76] |
| Daratumumab | 2019 | NTE NDMM |
IRs, URTI, diarrhea, constipation, peripheral edema, nausea, fatigue, asthenia, dyspnea, pyrexia, muscle spasms, and PSN | MAIA NTC02252172 |
[66][77][78] |
| Selinexor | 2019 | RRMM | Thrombocytopenia, fatigue, nausea, anemia, diarrhea, vomiting, hyponatremia, neutropenia, leukopenia, constipation, dyspnea, and URTI | STORM KCP-330-012 NCT02336815 |
[79][80] |
| Daratumumab | 2019 | TE NDMM |
IRs, PSN, constipation, asthenia, nausea, neutropenia, thrombocytopenia, peripheral edema, pyrexia and paresthesia | CASSIOPEIA NCT02541383 |
[81] |
NDMM: newly diagnosed multiple myeloma; RRMM: relapsed/refractory multiple myeloma; NTE: non-transplant-eligible; TE: transplant eligible; ORR: overall response rate; Vd: bortezomib, dexamethasone; PAN-Vd: panobinostat, bortezomib, dexamethasone; Rd: lenalidomide, dexamethasone; KRd: carfilzomib, lenalidomide, dexamethasone; DVd: daratumumab, bortezomib, dexamethasone; DRd: daratumumab, lenalidomide, dexamethasone; IRd: ixazomib, lenalidomide + dexamethasone; VTd: bortezomib, thalidomide, and dexamethasone; DVTd: daratumumab, bortezomib, thalidomide, dexamethasone; ERd: elotuzumab, lenalidomide, dexamethasone; Sd: selinexor, dexamethasone; m: months; PFS: progression-free survival; NR: Not Reached; URTI: Upper respiratory tract infection; PSN: Peripheral sensory neuropathy; IRs: Infusion reactions; VTE: venous thromboembolic events; CVE: Cardiovascular events, ARF: acute renal failure.
Treatment choices differ according to age, cytogenic abnormalities, and eligibility for transplantation. Maintenance therapy for standard-risk MM patients is lenalidomide. However, bortezomib is used as a maintenance therapy for high-risk ND patients who are determined to be eligible for ASCT. ND high-risk patients who are eligible for ASCT start with three to four cycles of VRd or three to four cycles of quadruplet regimen of daratumumab, bortezomib, lenalidomide, and dexamethasone (DVRd) [59].
2.1. Mechanism of Action of Proteasome Inhibitors
PIs kill myeloma cells through different pathways (Figure 3). Inhibition of proteosomes leads to the accumulation of ubiquitinated proteins that would otherwise be degraded in the proteosome. This leads to the accumulation of these proteins in the endoplasmic reticulum (ER), which in turn causes ER stress, which leads to ER stress-dependent apoptosis and activation of the Jun amino-terminal kinases (JNKs) pathway, increasing the Fas ligand, caspase 8, and caspase 3 [82][83]. Furthermore, mitochondrial injury occurs due to the direct effect of ubiquitinated proteins and the indirect effect of the ER stress that releases reactive oxygen species (ROS) [84]. The direct apoptosis effect of PIs can also occur through accumulation and phosphorylation of P53, which stimulates pro-apoptotic proteins such as Bcl-2-associated X protein (Bax), NADPH oxidase activator (NOXA), cytochrome-c release, and inhibition of the antiapoptotic protein Mcl-1 [85][86].
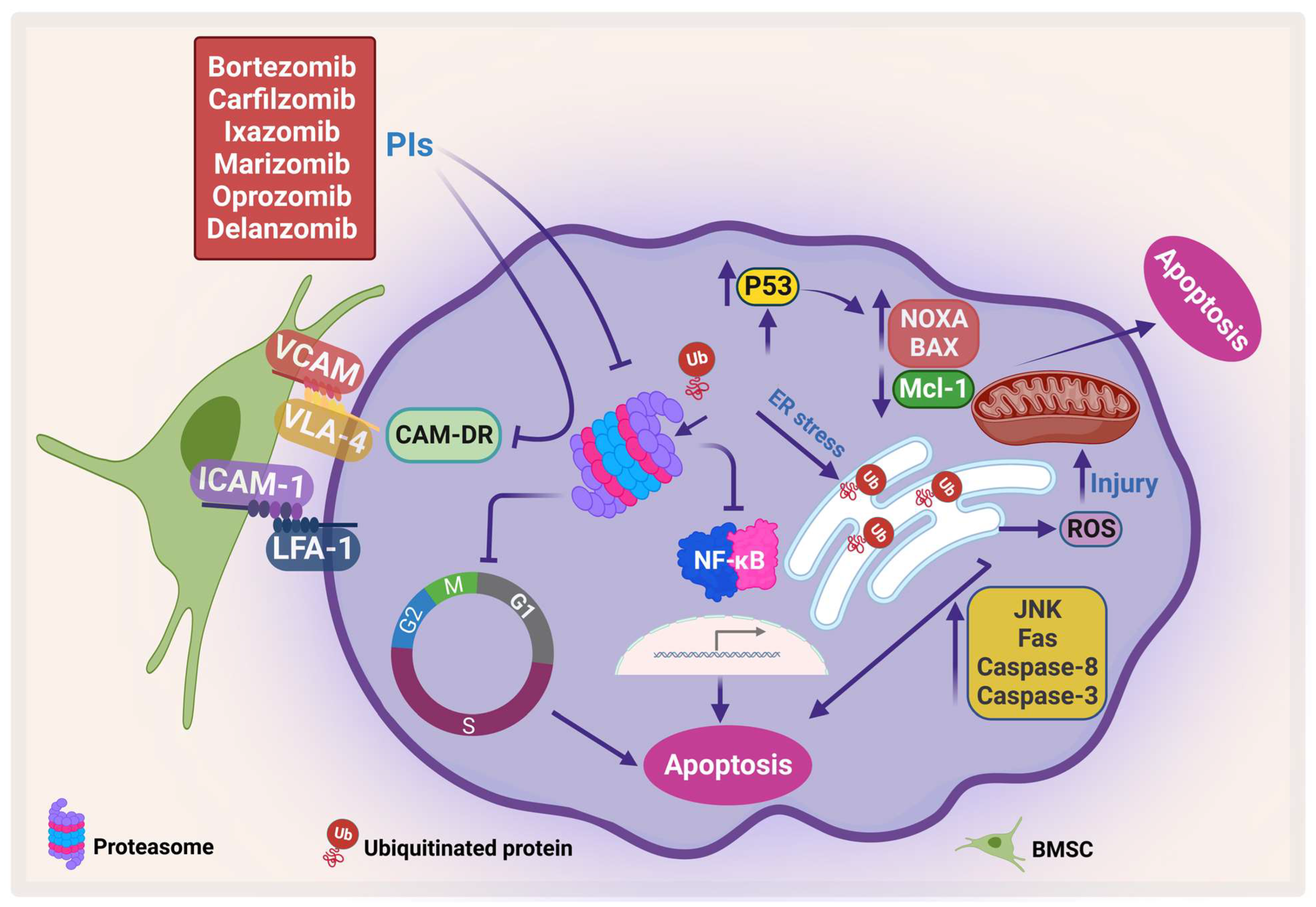
Figure 3. Proteasome inhibitors (PIs) induce apoptosis and inhibit CAM-DR. PIs induce protein accumulation, which leads to ER stress and activates JNK, which in turn activates caspase-8 and caspase-3, increases Fas, and generates ROS. In addition, PI enhances pro-apoptotic proteins, NOXA and BAX, and inhibits Mcl-1, which leads to apoptosis. Moreover, PI inhibits CAM-DR through inhibition of adhesion molecules (adapted from [86]). Created by Biorender.com.
Bortezomib is the first-generation FDA-approved PI that reversibly inhibits the chymotrypsin-like activity of the proteasomes [87]. Bortezomib has been shown to inhibit NF-κB, which in turn inhibits its downstream pathways and their products, including IL-6, vascular endothelial growth factor (VEGF), c-Myc, and cyclin D1 [88]. On the other hand, bortezomib has been shown to induce constitutive NF-κB activity which could be due to the difference in response among different cell clones [89][90] (reviewed in [86]). In addition, bortezomib ameliorates CAM-DR by inhibiting the expression of adhesion molecules such as VLA-4. Therefore, it resensitizes MM cells to treatment [91]. Even though introducing bortezomib has numerous benefits for patients, several side effects such as peripheral neuropathy may occur. The second-generation PI, carfilzomib, which does not cross the blood–brain barrier may have lower incidence of neuropathy. However, caution should be taken in using carfilzomib in elderly patients as it has shown cardiovascular side effects (reviewed at [92]). Similarly, ixazomib showed lower neurotoxicity than bortezomib as well as more efficacy in clinical trials [93].
2.2. Mechanism of Action of Immunomodulatory Drugs
IMiDs target some proteins for ubiquitination and proteasomal degradation through binding with cereblon ubiquitin ligase, forming an E3 ubiquitin ligase complex with DNA damage-binding protein 1, Cullin-4A, and RING box protein-1 (Figure 4). They target IKAROS family zinc finger 1 and 3 (IKZF1 and IKZF3), which are transcription factors that play an important role in lymphocyte biology. IKZF3 is an essential transcription factor in plasma cell development and therefore its degradation affects MM progression [94].
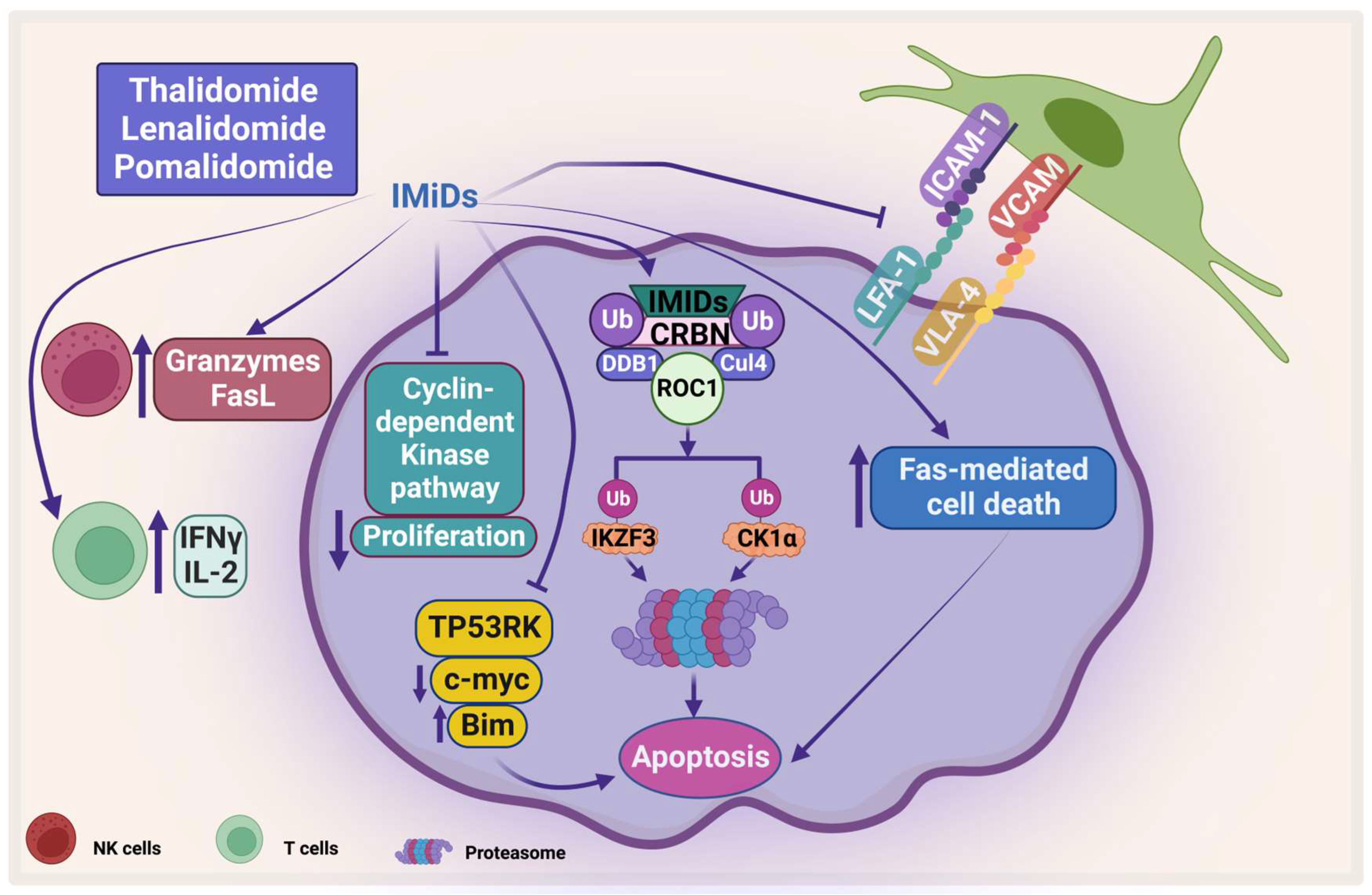
Figure 4. The direct and indirect effect of IMiDs on MM. IMiDs enhances apoptosis and inhibits MM proliferation through the cyclin-dependent pathway. IMiDs works indirectly through activating the immune cells. CRBN: Cereblon, DDB1: DNA damage-binding protein 1, CUL4: Cullin-4A, and ROC1: RING box protein-1. Created by Biorender.com.
Another protein that has been shown to be degraded by IMiDs is casein kinase 1 alpha (CK1α), which plays an important role in the pathogenesis of MM. CK is one of the serine/threonine kinases that is important in cell survival and has been shown to be important in different types of cancer including MM [95]. Manni et al. have shown that CK1α is overexpressed in most patients’ samples and its inhibition leads to apoptosis, a decrease in β-catenin and AKT expression, and an increase in p53 and p21 expression. In addition, the same group showed that CK1α inhibition enhances the cytotoxicity of bortezomib and lenalidomide on MM [96]. Interestingly, both CK1α and CK2 have been shown to sustain activation of important signaling pathways such as JAK/STAT, NF-κB, and PI3K/AKT [95][97][98]. IMiDs also inhibit the proliferation of PCs by inhibiting the cyclin-dependent kinase pathway through inducing P21 [99]. Moreover, IMiDs induce direct apoptosis in PCs by activation of Fas-mediated cell death [100][101]. Moreover, Hideshima et al. showed that IMiDs inhibit the kinase activity of p53-related protein kinase (TP53RK), which correlate negatively with MM patients’ survival. TP53RK phosphorylate serine 15 of p53, which in turn affects MM growth. The binding of IMiDs to TP53RK triggers apoptosis by inducing pro-apoptotic protein Bim. It also affects the metastasis of MM cells by inhibiting c-Myc protein [102].
The treatment of IMiDs helps in restoring the immune homeostasis. Several mechanisms have been proposed for IMiDs-mediated immune restoration, for example thalidomide stimulates the proliferation of T-cells and increases the secretion of interferon-gamma (IFN-γ) and interleukin-2 (IL-2) [103][104]. Along these lines, lenalidomide has been shown to stimulate T-cell-mediated cytotoxicity, induction of T-cell proliferative responses to allogeneic dendritic cells, and suppresses expression of programmed cell death protein-1 (PD-1) [105][106]. In addition, lenalidomide and pomalidomide suppress forkhead box P3 transcription factor and T regulatory cell expansion [107]. Moreover, lenalidomide enhances the expression of Fas ligand on NK cells and increases granzyme secretion which correlates to an increase in Ab-dependent cellular cytotoxicity (ADCC) [100]. Lenalidomide and pomalidomide inhibit the expression of adhesion molecules and inhibit the RANKL/osteoprotegerin ratio, which leads to inhibition of osteoclast formation [108].
2.3. Mechanism of Action of Histone Deacetylase Inhibitors
Dysregulation in epigenetics including histone acetylation has been shown in different types of cancer including MM [109][110]. Mithraprabhu et al. have shown that class I histone deacetylase (HDAC) is significantly upregulated in MM patients’ samples compared with normal PCs [111]. Moreover, upregulation of HDAC1 was correlated with poor prognosis and shorter OS [111].
Removal of the acetyl group from the lysine residue on histone by HDACs causes transcription repression (Figure 5). HDACs affect different proteins via deacetylation either directly or indirectly by affecting the function of the chaperone protein that is needed for their stabilization. HDACs cause hyperacetylation and therefore destabilization for the chaperone protein heat shock protein 90 (HSP90), which inhibits its association with CXCR4 leading to proteasomal degradation of CXCR4 in acute myeloid leukemia (AML) cells [112][113]. HDACs also cause degradation of protein phosphatase 3 catalytic subunit alpha (PPP3CA), which is overexpressed in MM, and patients show poor prognosis when it is overexpressed. As HDAC6 plays an important role in the aggresomal protein degradation, its inhibition significantly synergizes with proteasomal inhibition in MM [114]. Hideshima et al. have shown that a selective HDAC6 inhibitor increased the cytotoxicity of bortezomib in vitro and overcame its resistance through JNK activation and ER stress [115]. In 2015, panobinostat was approved for treatment of RRMM patients in combination with dexamethasone. HDAC inhibitors (HDACis) have been shown to affect the acetylation of histone and non-histone proteins; they therefore affect different cell processes including apoptosis, survival, angiogenesis, and the cell cycle [116][117].
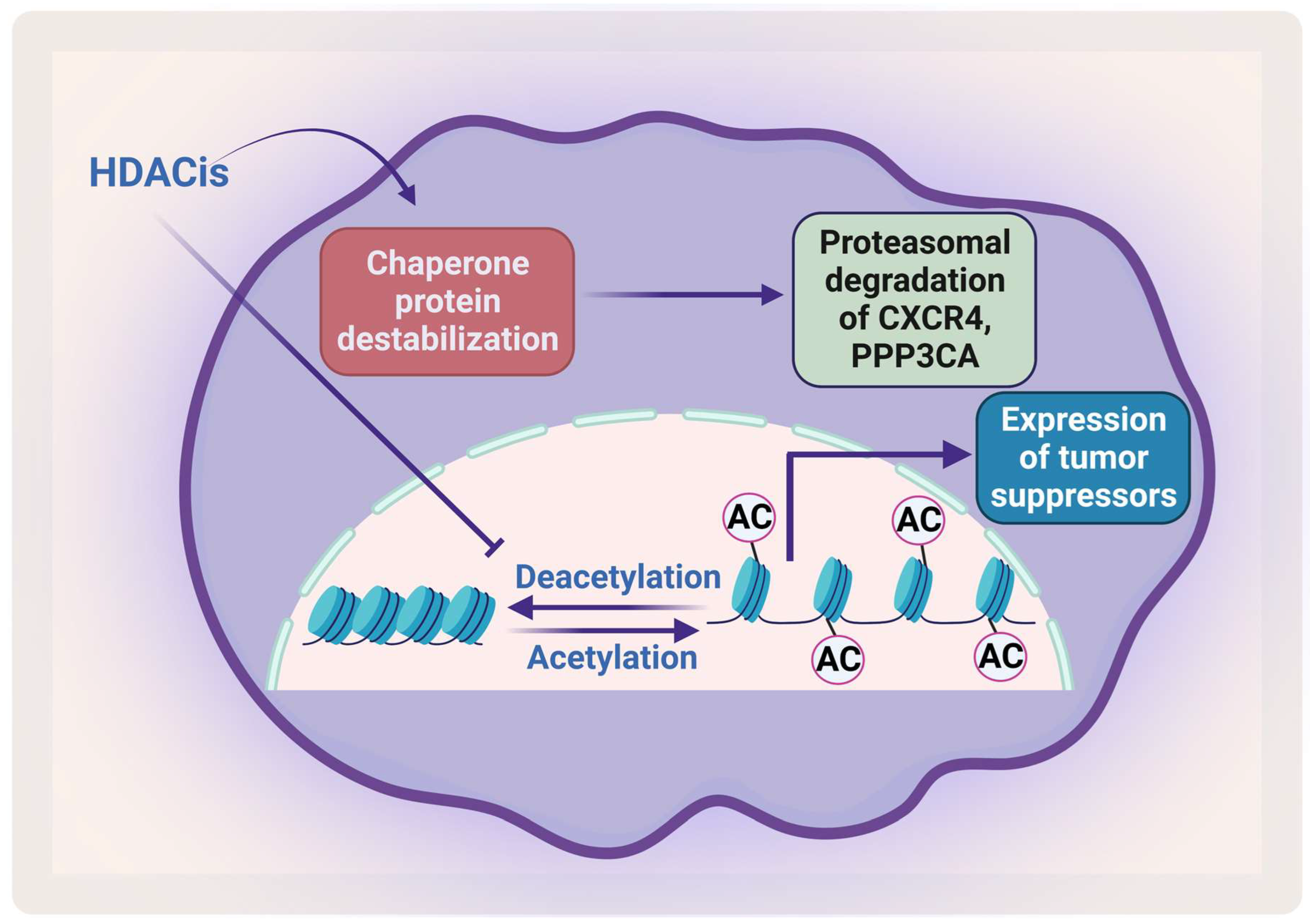
Figure 5. The effect of HDACis on MM. HDACis cause proteasomal degradation of CXCR4 and PPP3CA and enhance the expression of tumor suppressors. Created by Biorender.com.
References
- Kyle, R.A.; Durie, B.G.; Rajkumar, S.V.; Landgren, O.; Blade, J.; Merlini, G.; Kröger, N.; Einsele, H.; Vesole, D.H.; Dimopoulos, M.; et al. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia 2010, 24, 1121–1127.
- Atrash, S.; Robinson, M.; Slaughter, D.; Aneralla, A.; Brown, T.; Robinson, J.; Ndiaye, A.; Sprouse, C.; Zhang, Q.; Symanowski, J.T.; et al. Evolving changes in M-protein and hemoglobin as predictors for progression of smoldering multiple myeloma. Blood Cancer J. 2018, 8, 107.
- Rajkumar, S.V.; Landgren, O.; Mateos, M.V. Smoldering multiple myeloma. Blood 2015, 125, 3069–3075.
- Dimopoulos, M.A.; Terpos, E.; Chanan-Khan, A.; Leung, N.; Ludwig, H.; Jagannath, S.; Niesvizky, R.; Giralt, S.; Fermand, J.P.; Bladé, J.; et al. Renal impairment in patients with multiple myeloma: A consensus statement on behalf of the International Myeloma Working Group. J. Clin. Oncol. 2010, 28, 4976–4984.
- Mithraprabhu, S.; Khong, T.; Ramachandran, M.; Chow, A.; Klarica, D.; Mai, L.; Walsh, S.; Broemeling, D.; Marziali, A.; Wiggin, M.; et al. Circulating tumour DNA analysis demonstrates spatial mutational heterogeneity that coincides with disease relapse in myeloma. Leukemia 2017, 31, 1695–1705.
- Bergsagel, P.L.; Kuehl, W.M. Promiscuous Structural Variants Drive Myeloma Initiation and Progression. Blood Cancer Discov. 2020, 1, 221–223.
- Rajkumar, S.V. Multiple myeloma: 2022 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2022, 97, 1086–1107.
- Kumar, S.K.; Rajkumar, S.V. The multiple myelomas-current concepts in cytogenetic classification and therapy. Nat. Rev. Clin. Oncol 2018, 15, 409–421.
- Meads, M.B.; Hazlehurst, L.A.; Dalton, W.S. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin. Cancer Res. 2008, 14, 2519–2526.
- Bommert, K.; Bargou, R.C.; Stühmer, T. Signalling and survival pathways in multiple myeloma. Eur. J. Cancer 2006, 42, 1574–1580.
- Yosifov, D.Y.; Reufsteck, C.; Konstantinov, S.M.; Berger, M.R. Interleukin-6, osteopontin and Raf/MEK/ERK signaling modulate the sensitivity of human myeloma cells to alkylphosphocholines. Leuk. Res. 2012, 36, 764–772.
- van de Donk, N.W.; Lokhorst, H.M.; Bloem, A.C. Growth factors and antiapoptotic signaling pathways in multiple myeloma. Leukemia 2005, 19, 2177–2185.
- Ghobrial, I.M.; Detappe, A.; Anderson, K.C.; Steensma, D.P. The bone-marrow niche in MDS and MGUS: Implications for AML and MM. Nat. Rev. Clin. Oncol. 2018, 15, 219–233.
- Pawlyn, C.; Morgan, G.J. Evolutionary biology of high-risk multiple myeloma. Nat. Rev. Cancer 2017, 17, 543–556.
- Neumeister, P.; Schulz, E.; Pansy, K.; Szmyra, M.; Deutsch, A.J. Targeting the Microenvironment for Treating Multiple Myeloma. Int. J. Mol. Sci. 2022, 23, 7627.
- Di Marzo, L.; Desantis, V.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; Fumarulo, R.; Vacca, A.; Frassanito, M.A. Microenvironment drug resistance in multiple myeloma: Emerging new players. Oncotarget 2016, 7, 60698–60711.
- Mitsiades, C.S.; Mitsiades, N.S.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. The role of the bone microenvironment in the pathophysiology and therapeutic management of multiple myeloma: Interplay of growth factors, their receptors and stromal interactions. Eur. J. Cancer 2006, 42, 1564–1573.
- Uchiyama, H.; Barut, B.A.; Mohrbacher, A.F.; Chauhan, D.; Anderson, K.C. Adhesion of human myeloma-derived cell lines to bone marrow stromal cells stimulates interleukin-6 secretion. Blood 1993, 82, 3712–3720.
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348.
- Bleul, C.C.; Fuhlbrigge, R.C.; Casasnovas, J.M.; Aiuti, A.; Springer, T.A. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1). J. Exp. Med. 1996, 184, 1101–1109.
- Janssens, R.; Struyf, S.; Proost, P. The unique structural and functional features of CXCL12. Cell Mol. Immunol. 2018, 15, 299–311.
- Ma, Q.; Jones, D.; Borghesani, P.R.; Segal, R.A.; Nagasawa, T.; Kishimoto, T.; Bronson, R.T.; Springer, T.A. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 9448–9453.
- Gassmann, P.; Haier, J.; Schlüter, K.; Domikowsky, B.; Wendel, C.; Wiesner, U.; Kubitza, R.; Engers, R.; Schneider, S.W.; Homey, B.; et al. CXCR4 regulates the early extravasation of metastatic tumor cells in vivo. Neoplasia 2009, 11, 651–661.
- Hideshima, T.; Chauhan, D.; Hayashi, T.; Podar, K.; Akiyama, M.; Gupta, D.; Richardson, P.; Munshi, N.; Anderson, K.C. The biological sequelae of stromal cell-derived factor-1alpha in multiple myeloma. Mol. Cancer Ther. 2002, 1, 539–544.
- Hideshima, T.; Chauhan, D.; Schlossman, R.; Richardson, P.; Anderson, K.C. The role of tumor necrosis factor alpha in the pathophysiology of human multiple myeloma: Therapeutic applications. Oncogene 2001, 20, 4519–4527.
- Waldschmidt, J.M.; Simon, A.; Wider, D.; Müller, S.J.; Follo, M.; Ihorst, G.; Decker, S.; Lorenz, J.; Chatterjee, M.; Azab, A.K.; et al. CXCL12 and CXCR7 are relevant targets to reverse cell adhesion-mediated drug resistance in multiple myeloma. Br. J. Haematol. 2017, 179, 36–49.
- Liu, Y.; Liang, H.M.; Lv, Y.Q.; Tang, S.M.; Cheng, P. Blockade of SDF-1/CXCR4 reduces adhesion-mediated chemoresistance of multiple myeloma cells via interacting with interleukin-6. J. Cell Physiol. 2019, 234, 19702–19714.
- Sun, L.; Liu, L.; Liu, X.; Wang, Y.; Li, M.; Yao, L.; Yang, J.; Ji, G.; Guo, C.; Pan, Y.; et al. MGr1-Ag/37LRP induces cell adhesion-mediated drug resistance through FAK/PI3K and MAPK pathway in gastric cancer. Cancer Sci. 2014, 105, 651–659.
- Kobune, M.; Chiba, H.; Kato, J.; Kato, K.; Nakamura, K.; Kawano, Y.; Takada, K.; Takimoto, R.; Takayama, T.; Hamada, H.; et al. Wnt3/RhoA/ROCK signaling pathway is involved in adhesion-mediated drug resistance of multiple myeloma in an autocrine mechanism. Mol. Cancer Ther. 2007, 6, 1774–1784.
- Schmidmaier, R.; Baumann, P.; Simsek, M.; Dayyani, F.; Emmerich, B.; Meinhardt, G. The HMG-CoA reductase inhibitor simvastatin overcomes cell adhesion-mediated drug resistance in multiple myeloma by geranylgeranylation of Rho protein and activation of Rho kinase. Blood 2004, 104, 1825–1832.
- Shay, G.; Hazlehurst, L.; Lynch, C.C. Dissecting the multiple myeloma-bone microenvironment reveals new therapeutic opportunities. J. Mol. Med. 2016, 94, 21–35.
- Chong, P.S.Y.; Chng, W.J.; de Mel, S. STAT3: A Promising Therapeutic Target in Multiple Myeloma. Cancers 2019, 11, 731.
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746.
- Herrmann, A.; Kortylewski, M.; Kujawski, M.; Zhang, C.; Reckamp, K.; Armstrong, B.; Wang, L.; Kowolik, C.; Deng, J.; Figlin, R.; et al. Targeting Stat3 in the myeloid compartment drastically improves the in vivo antitumor functions of adoptively transferred T cells. Cancer Res. 2010, 70, 7455–7464.
- Kujawski, M.; Zhang, C.; Herrmann, A.; Reckamp, K.; Scuto, A.; Jensen, M.; Deng, J.; Forman, S.; Figlin, R.; Yu, H. Targeting STAT3 in adoptively transferred T cells promotes their in vivo expansion and antitumor effects. Cancer Res. 2010, 70, 9599–9610.
- Raje, N.S.; Bhatta, S.; Terpos, E. Role of the RANK/RANKL Pathway in Multiple Myeloma. Clin. Cancer Res. 2019, 25, 12–20.
- Liles, W.C.; Rodger, E.; Broxmeyer, H.E.; Dehner, C.; Badel, K.; Calandra, G.; Christensen, J.; Wood, B.; Price, T.H.; Dale, D.C. Augmented mobilization and collection of CD34+ hematopoietic cells from normal human volunteers stimulated with granulocyte-colony-stimulating factor by single-dose administration of AMD3100, a CXCR4 antagonist. Transfusion 2005, 45, 295–300.
- Flomenberg, N.; Devine, S.M.; Dipersio, J.F.; Liesveld, J.L.; McCarty, J.M.; Rowley, S.D.; Vesole, D.H.; Badel, K.; Calandra, G. The use of AMD3100 plus G-CSF for autologous hematopoietic progenitor cell mobilization is superior to G-CSF alone. Blood 2005, 106, 1867–1874.
- Bergsagel, D.E.; Sprague, C.C.; Austin, C.; Griffith, K.M. Evaluation of new chemotherapeutic agents in the treatment of multiple myeloma. IV. L-Phenylalanine mustard (NSC-8806). Cancer Chemother Rep. 1962, 21, 87–99.
- Hoogstraten, B.; Sheehe, P.R.; Cuttner, J.; Cooper, T.; Kyle, R.A.; Oberfield, R.A.; Townsend, S.R.; Harley, J.B.; Hayes, D.M.; Costa, G.; et al. Melphalan in multiple myeloma. Blood 1967, 30, 74–83.
- Gay, F.; Larocca, A.; Wijermans, P.; Cavallo, F.; Rossi, D.; Schaafsma, R.; Genuardi, M.; Romano, A.; Liberati, A.M.; Siniscalchi, A.; et al. Complete response correlates with long-term progression-free and overall survival in elderly myeloma treated with novel agents: Analysis of 1175 patients. Blood 2011, 117, 3025–3031.
- Palumbo, A.; Bringhen, S.; Caravita, T.; Merla, E.; Capparella, V.; Callea, V.; Cangialosi, C.; Grasso, M.; Rossini, F.; Galli, M.; et al. Oral melphalan and prednisone chemotherapy plus thalidomide compared with melphalan and prednisone alone in elderly patients with multiple myeloma: Randomised controlled trial. Lancet 2006, 367, 825–831.
- Osserman, E.F.; DiRe, L.B.; DiRe, J.; Sherman, W.H.; Hersman, J.A.; Storb, R. Identical twin marrow transplantation in multiple myeloma. Acta Haematol. 1982, 68, 215–223.
- Attal, M.; Harousseau, J.L.; Stoppa, A.M.; Sotto, J.J.; Fuzibet, J.G.; Rossi, J.F.; Casassus, P.; Maisonneuve, H.; Facon, T.; Ifrah, N.; et al. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. Intergroupe Francais du Myelome. N. Engl. J. Med. 1996, 335, 91–97.
- Twombly, R. First proteasome inhibitor approved for multiple myeloma. J. Natl. Cancer Inst. 2003, 95, 845.
- San Miguel, J.F.; Schlag, R.; Khuageva, N.K.; Dimopoulos, M.A.; Shpilberg, O.; Kropff, M.; Spicka, I.; Petrucci, M.T.; Palumbo, A.; Samoilova, O.S.; et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N. Engl. J. Med. 2008, 359, 906–917.
- Moreau, P.; Masszi, T.; Grzasko, N.; Bahlis, N.J.; Hansson, M.; Pour, L.; Sandhu, I.; Ganly, P.; Baker, B.W.; Jackson, S.R.; et al. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 374, 1621–1634.
- Rajkumar, S.V.; Blood, E.; Vesole, D.; Fonseca, R.; Greipp, P.R. Phase III clinical trial of thalidomide plus dexamethasone compared with dexamethasone alone in newly diagnosed multiple myeloma: A clinical trial coordinated by the Eastern Cooperative Oncology Group. J. Clin. Oncol. 2006, 24, 431–436.
- Services USDoHaH. Hematology/Oncology (Cancer) Approvals & Safety Notifications: Previous News Items; U.S. Food and Drug Administration. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/oncology-cancer-hematologic-malignancies-approval-notifications (accessed on 1 March 2023).
- Dimopoulos, M.; Spencer, A.; Attal, M.; Prince, H.M.; Harousseau, J.-L.; Dmoszynska, A.; Miguel, J.S.; Hellmann, A.; Facon, T.; Foà, R.; et al. Lenalidomide plus Dexamethasone for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2007, 357, 2123–2132.
- Orlowski, R.Z.; Nagler, A.; Sonneveld, P.; Bladé, J.; Hajek, R.; Spencer, A.; San Miguel, J.; Robak, T.; Dmoszynska, A.; Horvath, N.; et al. Randomized phase III study of pegylated liposomal doxorubicin plus bortezomib compared with bortezomib alone in relapsed or refractory multiple myeloma: Combination therapy improves time to progression. J. Clin. Oncol. 2007, 25, 3892–3901.
- Richardson, P.G.; Xie, W.; Mitsiades, C.; Chanan-Khan, A.A.; Lonial, S.; Hassoun, H.; Avigan, D.E.; Oaklander, A.L.; Kuter, D.J.; Wen, P.Y.; et al. Single-agent bortezomib in previously untreated multiple myeloma: Efficacy, characterization of peripheral neuropathy, and molecular correlations with response and neuropathy. J. Clin. Oncol. 2009, 27, 3518–3525.
- Sánchez-Ortega, I.; Querol, S.; Encuentra, M.; Ortega, S.; Serra, A.; Sanchez-Villegas, J.M.; Grifols, J.R.; Pujol-Balaguer, M.M.; Pujol-Bosch, M.; Martí, J.M.; et al. Plerixafor in patients with lymphoma and multiple myeloma: Effectiveness in cases with very low circulating CD34+ cell levels and preemptive intervention vs remobilization. Bone Marrow Transpl. 2015, 50, 34–39.
- Administration USFaD. Carfilzomib. Available online: http://wayback.archive-it.org/7993/20170113081106/http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm312945.htm (accessed on 1 March 2023).
- Richardson, P.G.; Siegel, D.S.; Vij, R.; Hofmeister, C.C.; Baz, R.; Jagannath, S.; Chen, C.; Lonial, S.; Jakubowiak, A.; Bahlis, N.; et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: A randomized phase 2 study. Blood 2014, 123, 1826–1832.
- Kumar, S.K.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Zeldenrust, S.R.; Dingli, D.; Russell, S.J.; Lust, J.A.; et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008, 111, 2516–2520.
- Attal, M.; Lauwers-Cances, V.; Marit, G.; Caillot, D.; Moreau, P.; Facon, T.; Stoppa, A.M.; Hulin, C.; Benboubker, L.; Garderet, L.; et al. Lenalidomide Maintenance after Stem-Cell Transplantation for Multiple Myeloma. N. Engl. J. Med. 2012, 366, 1782–1791.
- Durie, B.G.M.; Hoering, A.; Abidi, M.H.; Rajkumar, S.V.; Epstein, J.; Kahanic, S.P.; Thakuri, M.; Reu, F.; Reynolds, C.M.; Sexton, R.; et al. Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777): A randomised, open-label, phase 3 trial. Lancet 2017, 389, 519–527.
- Rajkumar, S.V.; Kumar, S. Multiple myeloma current treatment algorithms. Blood Cancer J. 2020, 10, 94.
- Miguel, J.S.; Weisel, K.; Moreau, P.; Lacy, M.; Song, K.; Delforge, M.; Karlin, L.; Goldschmidt, H.; Banos, A.; Oriol, A.; et al. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): A randomised, open-label, phase 3 trial. Lancet Oncol. 2013, 14, 1055–1066.
- Sanofi. Sanofi: FDA Approves Sarclisa® (isatuximab-irfc) for Patients with Relapsed Refractory Multiple Myeloma. Available online: https://www.sanofi.com/en/media-room/press-releases/2020/2020-03-02-18-51-16-1993727 (accessed on 1 March 2023).
- Attal, M.; Richardson, P.G.; Rajkumar, S.V.; San-Miguel, J.; Beksac, M.; Spicka, I.; Leleu, X.; Schjesvold, F.; Moreau, P.; Dimopoulos, M.A.; et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): A randomised, multicentre, open-label, phase 3 study. Lancet 2019, 394, 2096–2107.
- Richardson, P.G.; Laubach, J.P.; Munshi, N.C.; Anderson, K.C. Early or delayed transplantation for multiple myeloma in the era of novel therapy: Does one size fit all? Hematol. Am. Soc. Hematol. Educ. Program. 2014, 2014, 255–261.
- Cavo, M.; San-Miguel, J.; Usmani, S.Z.; Weisel, K.; Dimopoulos, M.A.; Avet-Loiseau, H.; Paiva, B.; Bahlis, N.J.; Plesner, T.; Hungria, V.; et al. Prognostic value of minimal residual disease negativity in myeloma: Combined analysis of POLLUX, CASTOR, ALCYONE, and MAIA. Blood 2022, 139, 835–844.
- Facon, T.; Kumar, S.K.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab, lenalidomide, and dexamethasone versus lenalidomide and dexamethasone alone in newly diagnosed multiple myeloma (MAIA): Overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 1582–1596.
- Kapoor, P.; Rajkumar, S.V. MAIA under the microscope-bringing trial design into focus. Nat. Rev. Clin. Oncol. 2019, 16, 339–340.
- Mateos, M.V.; Dimopoulos, M.A.; Cavo, M.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; Kaplan, P.; et al. Daratumumab plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. N. Engl. J. Med. 2018, 378, 518–528.
- Moreau, P.; Dimopoulos, M.A.; Mikhael, J.; Yong, K.; Capra, M.; Facon, T.; Hajek, R.; Špička, I.; Baker, R.; Kim, K.; et al. Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma (IKEMA): A multicentre, open-label, randomised phase 3 trial. Lancet 2021, 397, 2361–2371.
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Günther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Overall survival of patients with relapsed multiple myeloma treated with panobinostat or placebo plus bortezomib and dexamethasone (the PANORAMA 1 trial): A randomised, placebo-controlled, phase 3 trial. Lancet Haematol. 2016, 3, e506–e515.
- Dimopoulos, M.A.; Stewart, A.K.; Masszi, T.; Špička, I.; Oriol, A.; Hájek, R.; Rosiñol, L.; Siegel, D.; Mihaylov, G.G.; Goranova-Marinova, V.; et al. Carfilzomib, lenalidomide, and dexamethasone in patients with relapsed multiple myeloma categorised by age: Secondary analysis from the phase 3 ASPIRE study. Br. J. Haematol. 2017, 177, 404–413.
- Lonial, S.; Weiss, B.M.; Usmani, S.Z.; Singhal, S.; Chari, A.; Bahlis, N.J.; Belch, A.; Krishnan, A.; Vescio, R.A.; Mateos, M.V.; et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): An open-label, randomised, phase 2 trial. Lancet 2016, 387, 1551–1560.
- Dimopoulos, M.A.; Lonial, S.; White, D.; Moreau, P.; Weisel, K.; San-Miguel, J.; Shpilberg, O.; Grosicki, S.; Špička, I.; Walter-Croneck, A.; et al. Elotuzumab, lenalidomide, and dexamethasone in RRMM: Final overall survival results from the phase 3 randomized ELOQUENT-2 study. Blood Cancer J. 2020, 10, 91.
- Dimopoulos, M.A.; Lonial, S.; White, D.; Moreau, P.; Palumbo, A.; San-Miguel, J.; Shpilberg, O.; Anderson, K.; Grosicki, S.; Spicka, I.; et al. Elotuzumab plus lenalidomide/dexamethasone for relapsed or refractory multiple myeloma: ELOQUENT-2 follow-up and post-hoc analyses on progression-free survival and tumour growth. Br. J. Haematol. 2017, 178, 896–905.
- Bahlis, N.J.; Dimopoulos, M.A.; White, D.J.; Benboubker, L.; Cook, G.; Leiba, M.; Ho, P.J.; Kim, K.; Takezako, N.; Moreau, P.; et al. Daratumumab plus lenalidomide and dexamethasone in relapsed/refractory multiple myeloma: Extended follow-up of POLLUX, a randomized, open-label, phase 3 study. Leukemia 2020, 34, 1875–1884.
- Spencer, A.; Lentzsch, S.; Weisel, K.; Avet-Loiseau, H.; Mark, T.M.; Spicka, I.; Masszi, T.; Lauri, B.; Levin, M.D.; Bosi, A.; et al. Daratumumab plus bortezomib and dexamethasone versus bortezomib and dexamethasone in relapsed or refractory multiple myeloma: Updated analysis of CASTOR. Haematologica 2018, 103, 2079–2087.
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766.
- Facon, T.; Kumar, S.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. N. Engl. J. Med. 2019, 380, 2104–2115.
- Auner, H.W.; Gavriatopoulou, M.; Delimpasi, S.; Simonova, M.; Spicka, I.; Pour, L.; Dimopoulos, M.A.; Kriachok, I.; Pylypenko, H.; Leleu, X.; et al. Effect of age and frailty on the efficacy and tolerability of once-weekly selinexor, bortezomib, and dexamethasone in previously treated multiple myeloma. Am. J. Hematol. 2021, 96, 708–718.
- Vogl, D.T.; Dingli, D.; Cornell, R.F.; Huff, C.A.; Jagannath, S.; Bhutani, D.; Zonder, J.; Baz, R.; Nooka, A.; Richter, J.; et al. Selective Inhibition of Nuclear Export With Oral Selinexor for Treatment of Relapsed or Refractory Multiple Myeloma. J. Clin. Oncol. 2018, 36, 859–866.
- Chari, A.; Vogl, D.T.; Gavriatopoulou, M.; Nooka, A.K.; Yee, A.J.; Huff, C.A.; Moreau, P.; Dingli, D.; Cole, C.; Lonial, S.; et al. Oral Selinexor-Dexamethasone for Triple-Class Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 381, 727–738.
- Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Béné, M.C.; Broijl, A.; Caillon, H.; Caillot, D.; et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): A randomised, open-label, phase 3 study. Lancet 2019, 394, 29–38.
- Meriin, A.B.; Gabai, V.L.; Yaglom, J.; Shifrin, V.I.; Sherman, M.Y. Proteasome inhibitors activate stress kinases and induce Hsp72. Diverse effects on apoptosis. J. Biol. Chem. 1998, 273, 6373–6379.
- Tani, E.; Kitagawa, H.; Ikemoto, H.; Matsumoto, T. Proteasome inhibitors induce Fas-mediated apoptosis by c-Myc accumulation and subsequent induction of FasL message in human glioma cells. FEBS Lett. 2001, 504, 53–58.
- Ling, Y.H.; Liebes, L.; Zou, Y.; Perez-Soler, R. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic response to Bortezomib, a novel proteasome inhibitor, in human H460 non-small cell lung cancer cells. J. Biol. Chem. 2003, 278, 33714–33723.
- Hideshima, T.; Mitsiades, C.; Akiyama, M.; Hayashi, T.; Chauhan, D.; Richardson, P.; Schlossman, R.; Podar, K.; Munshi, N.C.; Mitsiades, N.; et al. Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341. Blood 2003, 101, 1530–1534.
- Ito, S. Proteasome Inhibitors for the Treatment of Multiple Myeloma. Cancers 2020, 12, 265.
- Thibaudeau, T.A.; Smith, D.M. A Practical Review of Proteasome Pharmacology. Pharmacol. Rev. 2019, 71, 170–197.
- Hideshima, T.; Richardson, P.; Chauhan, D.; Palombella, V.J.; Elliott, P.J.; Adams, J.; Anderson, K.C. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001, 61, 3071–3076.
- Hideshima, T.; Ikeda, H.; Chauhan, D.; Okawa, Y.; Raje, N.; Podar, K.; Mitsiades, C.; Munshi, N.C.; Richardson, P.G.; Carrasco, R.D.; et al. Bortezomib induces canonical nuclear factor-kappaB activation in multiple myeloma cells. Blood 2009, 114, 1046–1052.
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.J.; Van Wier, S.; Tiedemann, R.; Shi, C.X.; Sebag, M.; et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 2007, 12, 131–144.
- Noborio-Hatano, K.; Kikuchi, J.; Takatoku, M.; Shimizu, R.; Wada, T.; Ueda, M.; Nobuyoshi, M.; Oh, I.; Sato, K.; Suzuki, T.; et al. Bortezomib overcomes cell-adhesion-mediated drug resistance through downregulation of VLA-4 expression in multiple myeloma. Oncogene 2009, 28, 231–242.
- Groen, K.; van de Donk, N.; Stege, C.; Zweegman, S.; Nijhof, I.S. Carfilzomib for relapsed and refractory multiple myeloma. Cancer Manag. Res. 2019, 11, 2663–2675.
- Sana, M.K.; Abdullah, S.M.; Javed, S.; Ehsan, H.; Faizan, U.; Khalid, F.; Jaan, A.; Tayyeb, M.; Abdullah, S.; Anwer, F. Efficacy of Ixazomib and Bortezomib with Lenalidomide Combination Regimens for Multiple Myeloma: A Systematic Review. Blood 2020, 136, 40–41.
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305.
- Manni, S.; Carrino, M.; Piazza, F. Role of protein kinases CK1α and CK2 in multiple myeloma: Regulation of pivotal survival and stress-managing pathways. J. Hematol. Oncol. 2017, 10, 157.
- Manni, S.; Carrino, M.; Manzoni, M.; Gianesin, K.; Nunes, S.C.; Costacurta, M.; Tubi, L.Q.; Macaccaro, P.; Taiana, E.; Cabrelle, A.; et al. Inactivation of CK1α in multiple myeloma empowers drug cytotoxicity by affecting AKT and β-catenin survival signaling pathways. Oncotarget 2017, 8, 14604–14619.
- Zheng, Y.; Qin, H.; Frank, S.J.; Deng, L.; Litchfield, D.W.; Tefferi, A.; Pardanani, A.; Lin, F.T.; Li, J.; Sha, B.; et al. A CK2-dependent mechanism for activation of the JAK-STAT signaling pathway. Blood 2011, 118, 156–166.
- Yu, M.; Yeh, J.; Van Waes, C. Protein kinase casein kinase 2 mediates inhibitor-kappaB kinase and aberrant nuclear factor-kappaB activation by serum factor(s) in head and neck squamous carcinoma cells. Cancer Res. 2006, 66, 6722–6731.
- Schafer, P.H.; Gandhi, A.K.; Zhang, L.-H.; Kang, J.; Capone, L.; Parton, S.; Wu, L.; Bartlett, B. Opposing Effects of Dexamethasone on Lenalidomide Activity in Multiple Myeloma: Additive/Synergistic Effects on Anti-Proliferative Activity on Myeloma Cells and Antagonistic Effects on Immune Function. Blood 2008, 112, 2761.
- Wu, L.; Adams, M.; Carter, T.; Chen, R.; Muller, G.; Stirling, D.; Schafer, P.; Bartlett, J.B. lenalidomide enhances natural killer cell and monocyte-mediated antibody-dependent cellular cytotoxicity of rituximab-treated CD20+ tumor cells. Clin. Cancer Res. 2008, 14, 4650–4657.
- Quach, H.; Ritchie, D.; Stewart, A.K.; Neeson, P.; Harrison, S.; Smyth, M.J.; Prince, H.M. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia 2010, 24, 22–32.
- Hideshima, T.; Cottini, F.; Nozawa, Y.; Seo, H.S.; Ohguchi, H.; Samur, M.K.; Cirstea, D.; Mimura, N.; Iwasawa, Y.; Richardson, P.G.; et al. p53-related protein kinase confers poor prognosis and represents a novel therapeutic target in multiple myeloma. Blood 2017, 129, 1308–1319.
- Haslett, P.A.; Corral, L.G.; Albert, M.; Kaplan, G. Thalidomide costimulates primary human T lymphocytes, preferentially inducing proliferation, cytokine production, and cytotoxic responses in the CD8+ subset. J. Exp. Med. 1998, 187, 1885–1892.
- Davies, F.E.; Raje, N.; Hideshima, T.; Lentzsch, S.; Young, G.; Tai, Y.T.; Lin, B.; Podar, K.; Gupta, D.; Chauhan, D.; et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood 2001, 98, 210–216.
- Luptakova, K.; Glotzbecker, B.; Mills, H.; Stroopinsky, D.; Vasir, B.; Rosenblatt, J.; Kufe, D.; Avigan, D. Lenalidomide Decreases PD-1 Expression, Depletes Regulatory T-Cells and Improves Cellular Response to a Multiple Myeloma/Dendritic Cell Fusion Vaccine In Vitro. Blood 2010, 116, 492.
- Luptakova, K.; Rosenblatt, J.; Glotzbecker, B.; Mills, H.; Stroopinsky, D.; Kufe, T.; Vasir, B.; Arnason, J.; Tzachanis, D.; Zwicker, J.I.; et al. Lenalidomide enhances anti-myeloma cellular immunity. Cancer Immunol. Immunother. 2013, 62, 39–49.
- Galustian, C.; Meyer, B.; Labarthe, M.-C.; Dredge, K.; Klaschka, D.; Henry, J.; Todryk, S.; Chen, R.; Muller, G.; Stirling, D.; et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol. Immunother. 2009, 58, 1033–1045.
- Bolzoni, M.; Storti, P.; Bonomini, S.; Todoerti, K.; Guasco, D.; Toscani, D.; Agnelli, L.; Neri, A.; Rizzoli, V.; Giuliani, N. Immunomodulatory drugs lenalidomide and pomalidomide inhibit multiple myeloma-induced osteoclast formation and the RANKL/OPG ratio in the myeloma microenvironment targeting the expression of adhesion molecules. Exp. Hematol. 2013, 41, 387–397.e381.
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018, 132, 587–597.
- Dutta, R.; Tiu, B.; Sakamoto, K.M. CBP/p300 acetyltransferase activity in hematologic malignancies. Mol. Genet. Metab. 2016, 119, 37–43.
- Mithraprabhu, S.; Kalff, A.; Chow, A.; Khong, T.; Spencer, A. Dysregulated Class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics 2014, 9, 1511–1520.
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39.
- Mandawat, A.; Fiskus, W.; Buckley, K.M.; Robbins, K.; Rao, R.; Balusu, R.; Navenot, J.M.; Wang, Z.X.; Ustun, C.; Chong, D.G.; et al. Pan-histone deacetylase inhibitor panobinostat depletes CXCR4 levels and signaling and exerts synergistic antimyeloid activity in combination with CXCR4 antagonists. Blood 2010, 116, 5306–5315.
- Hideshima, T.; Bradner, J.E.; Wong, J.; Chauhan, D.; Richardson, P.; Schreiber, S.L.; Anderson, K.C. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Natl. Acad. Sci. USA 2005, 102, 8567–8572.
- Hideshima, T.; Qi, J.; Paranal, R.M.; Tang, W.; Greenberg, E.; West, N.; Colling, M.E.; Estiu, G.; Mazitschek, R.; Perry, J.A.; et al. Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proc. Natl. Acad. Sci. USA 2016, 113, 13162–13167.
- Kaufman, J.L.; Fabre, C.; Lonial, S.; Richardson, P.G. Histone deacetylase inhibitors in multiple myeloma: Rationale and evidence for their use in combination therapy. Clin. Lymphoma Myeloma Leuk. 2013, 13, 370–376.
- Imai, Y.; Hirano, M.; Kobayashi, M.; Futami, M.; Tojo, A. HDAC Inhibitors Exert Anti-Myeloma Effects through Multiple Modes of Action. Cancers 2019, 11, 475.
More
Information
Subjects:
Hematology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.5K
Revisions:
2 times
(View History)
Update Date:
17 Mar 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No