Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nicola Perrotti | -- | 2813 | 2023-03-16 14:51:48 | | | |
| 2 | Lindsay Dong | + 5 word(s) | 2818 | 2023-03-17 04:59:30 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
D’antona, L.; Amato, R.; Brescia, C.; Rocca, V.; Colao, E.; Iuliano, R.; Blazer-Yost, B.L.; Perrotti, N. Kinase Inhibitors in Genetic Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/42274 (accessed on 27 June 2026).
D’antona L, Amato R, Brescia C, Rocca V, Colao E, Iuliano R, et al. Kinase Inhibitors in Genetic Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/42274. Accessed June 27, 2026.
D’antona, Lucia, Rosario Amato, Carolina Brescia, Valentina Rocca, Emma Colao, Rodolfo Iuliano, Bonnie L. Blazer-Yost, Nicola Perrotti. "Kinase Inhibitors in Genetic Diseases" Encyclopedia, https://encyclopedia.pub/entry/42274 (accessed June 27, 2026).
D’antona, L., Amato, R., Brescia, C., Rocca, V., Colao, E., Iuliano, R., Blazer-Yost, B.L., & Perrotti, N. (2023, March 16). Kinase Inhibitors in Genetic Diseases. In Encyclopedia. https://encyclopedia.pub/entry/42274
D’antona, Lucia, et al. "Kinase Inhibitors in Genetic Diseases." Encyclopedia. Web. 16 March, 2023.
Copy Citation
Gene variants may have functional consequences on the protein product. The variants can be classified as loss of function (LOF) when the protein function is reduced or lost and gain of function (GOF) when the protein function is enhanced or a new function is acquired. Kinase inhibitors have been used for many years in tumor therapy, and new monoclonal antibodies—as well as small inhibitory molecules with different indications in tumor therapy—are approved every year by the regulatory agencies. human epidermal growth factor receptor 2 (HER2) inhibitors are currently used in HER2-positive carcinomas, including breast, colon, and non-small-cell lung (NSCLC) cancers, Abl inhibitors are used in chronic myeloid leukemia.
kinase inhibitors
genetics
RAS pathway
mTOR pathway
Wnt pathway
1. Tuberous Sclerosis Complex
TSC is a disease characterized by autosomal dominant inheritance and has an incidence of 1 in 6000–10,000 individuals [1]. It is involved in the development of very different pathologies that involve the brain, heart, kidneys, and skin.
1.1. Clinical Manifestation
Neurological manifestations represent the most important cause of impairment in most patients. These include epilepsy, present in 90% of patients [2]; cortical tubers; subependymal nodules; giant cell astrocytomas; intellectual disability; autistic spectrum disorder; and behavioral problems [3][4].
Most patients with epilepsy have seizures in the first year of age [5]. Some studies have speculated that the seizures originate from cortical tubers, but the mechanism is still being studied [6]. Long-term damage depends on the age of onset and the severity of seizures [7][8].
The cortical tuber, from which the disease takes its name, is a focal malformation present in 80–90% of patients. It is due to a failure of cell differentiation with neuronal migration during neurological development [9].
1.2. Genetics of TSC
TSC is caused by mutations in two genes, the TSC1 gene located on chromosome 9 (9q34), which encodes the hamartina protein, and TSC2 located on chromosome 16 (16p13.3), which encodes the tuberin protein [10][11]. TSC1 mutations have been identified in ~10–20% of patients, whereas TSC2 mutations have been identified in ~70–90% of patients clinically diagnosed with TSC [12][13]. The proteins produced by these two genes physically interact with high affinity to form a heterotrimeric complex, termed the TSC protein complex, with the TBC1 domain family member 7 (TBC1D7), [14][15] and act on the signaling pathway of mTORC1 (rapamycin complex 1), a serine/threonine kinase involved in many cellular processes, such as cell growth, proliferation, and response to extracellular stress [16][17][18]. The major driver of the cellular hyperplasia and tissue dysplasia seen in TSC is the overactivation of the mTORC1 signaling pathway. Under normal conditions, growth factors stimulate the PI3K (phosphatidylinositol 3-kinase) and Ras–MAPK (mitogen-activated protein kinase) pathways, thereby inhibiting the TSC protein complex and activating mTOR signaling (Figure 1) [19][20]. mTOR acts through two complexes, mTORC1 and mTORC2. The loss of function of TSC genes results in an increase in mTORC1 and a decrease in mTORC2 signaling [21]. The mechanism by which this occurs is not clear; it is probably due to loss of direct binding of the amartin/tuberin complex to mTORC2. Alternatively, increasing mTORC1 activates a negative feedback loop from p70S6K inhibiting IRS-1 by inhibiting the PI3-kinase-dependent activation of mTORC2.

Figure 1. The figure shows how the mTORC1 pathway is involved in the development of TSC. The figure shows the genes involved in the development of TSC and the pathway activated by different proteins. In particular, mTORC1 may contribute to the symptoms of TSC when hyperactivated by the malfunction of the TBC1D7 tuberin and hamartin complex. Two of the FDA-approved kinase inhibitors for the treatment of TSC are also shown in the figure.
1.3. Inhibition of mTOR
The mTOR inhibitors in use today are sirolimus and everolimus. The former is also known as rapamycin and has been obtained from soil samples from Easter Island [22]. Everolimus is a sirolimus derivative obtained via the addition of an ethyl ester group. Both compounds induce allosteric dissociation of the cofactor rapTOR (TOR regulation-related protein) that is essential for mTOR function [23]. TSC-associated epilepsy does not respond well to treatment with traditional antiepileptics. Some trials show that mTOR inhibition can reduce the frequency of epileptic manifestations and reduce the volume of tumors and nodules [24][25].
2. RASopathies
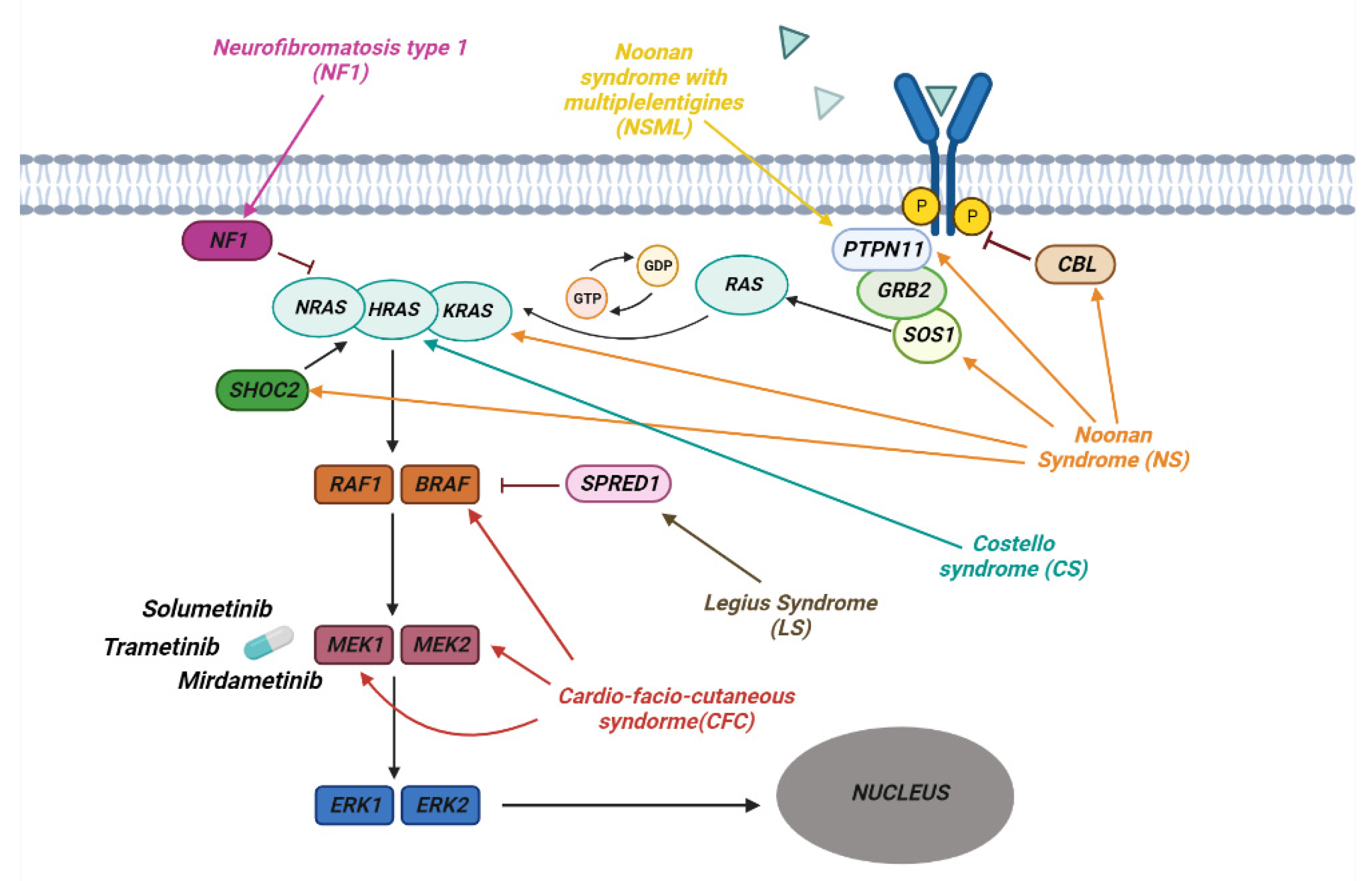
RASopathies affect about 1 in 1000 individuals and are part of a heterogeneous group of diseases in which there is a germline mutation in the genes that code for components and regulators of the Ras/MAPK Pathway (Figure 2). This is one of the best-studied and characterized pathways and is involved in the regulation of growth, cell cycle differentiation, and normal mammalian development.
Figure 2. The pathway of RAS/MAPK in RASopathies. The figure shows how the various genes of the Ras/MAPK pathway are involved in the development of the different syndromes associated with RASopathies. Shown are some of the MERK1/2 kinase inhibitors used to treat RASopathies, some of which are FDA-approved.
2.1. Clinical Manifestation
These pathologies each have their own peculiar phenotype but have common characteristics, such as cardiac malformations, craniofacial dysmorphologies, skin, ocular and musculoskeletal abnormalities, hypotonia, and predisposition to tumors [26].
2.2. Neurofibromatosis
The first to be identified was Neurofibromatosis type 1 (NF1). This is an autosomal dominant disease that affects 1 in 3000 individuals and is present in one of the parents in 50% of cases, whereas it is the result of a de novo mutation in the remaining 50%. NF1 is characterized by the development of progressive benign and malignant tumors of the central and peripheral nervous system [27]. Diagnosis is mainly based on the presence of café-au-lait maculae, intertriginous freckling, neurofibromas and plexiform neurofibromas, iris Lisch nodules, osseous dysplasia, optic pathway glioma, and other symptoms common to RASophaties in general. The most severe and frequent clinical manifestations in type 1 neurofibromatosis are plexiform neurofibromas (PNFs), low-grade gliomas, and other peripheral nerve sheath tumors. PNFs occurs in 20–50% of patients with NF1 [28]. These are tumors of the peripheral nerves that are generally benign but can cause complications such as disfigurement and pain [29][30]. PNFs are diagnosed in early childhood and may need to undergo surgical excision, although the procedure can be complicated by factors such as proximity to nerves and wide vascularity.
2.3. Noonan Syndrome
Noonan syndrome (NS) is one of the most common among RASopathies, in most cases it is inherited in an autosomal dominant fashion, with a variable expression.74,75 Clinical diagnosis is based on peculiar facial characteristics, short stature, cardiac defects—such as pulmonary stenosis or hypetrophic cardiomyopathy—and variable degrees of development delay. Noonan syndrome clinically overlaps with other RASopathies that are the consequence of genetic defects in the same RAS pathway.
In Noonan syndrome with multiple lentigines (NSML)—formerly known as Leopard syndrome, Costello Syndrome (CS), and Cardio-facio-cutaneous syndrome (CFC)—most of the features of Noonan syndrome are associated with severe mental retardation, severe feeding difficulties, myopathy, facial papillomata, and warts, particularly in the nasolabial area, loose skin on the hands and feet, with hyperkeratotic palms and soles.
2.4. Genetics of RASopathies
On the basis of the mutated gene of the Ras/MAPK pathway, RASopathies are classified into different groups. Some syndromes are due to the mutation of a single gene in this pathway, while others can arise as a result of the mutation of several genes.
NF1 is caused by a mutation in the NF1 gene encoding the neurofibromin which is RasGAP, a GTPase-activating protein, that regulates Ras negatively. This mutation results in a loss of protein function followed by increased Ras activity that explains the predisposition to the development of malignant tumors observed in NF1 patients [31].
Noonan Syndrome is due to germline pathogenic variants involving genes such as PTPN11, SOS1, KRAS, NRAS, SHOC2, CBL and MAP2K1 [32]. The most frequent mutations are in the PTPN11, SOS1, and KRAS genes. These are generally de novo missense mutations that, once established in the germline, can be inherited in an autosomal dominant fashion. It is generally believed that missense mutations in the PTPN11 gene coding for the SHP-2 tyrosine phosphatase cause a gain of function in the enzymatic activity [33] and that active SHP-2 positively regulates cell growth and differentiation by promoting activation of the Ras–MAPK pathway (Figure 2) [34]. Mutations involving the SOS1 gene, a member of the RAS GEF family important in the conversion of GDP into GTP, lead to the activation of RAS [35][36]. The most frequent mutations of this gene occur in the codons that code for the genes involved in the maintenance of its self-inhibited form, leading to an increase in the activity of SOS1 and a continuous activation of RAS [36]. Other frequent mutations that lead to the onset of this syndrome are those affecting KRAS, which lead to a reduction in the hydrolysis of GTP with accumulation of the active form of KRAS [37][38]. Other rarer mutations, leading to the development of Noonan Syndrome, have been observed in the BRAF, SHOC2, and CBL genes [39][40][41].
CS is an autosomal dominant disease characterized by germline mutations of the HRAS gene, located on chromosome 11p15.5. One of the most common variants is pGly12Ser, which leads to greater activation of HRAS following a reduction in GAP-induced GTPase activity [42][43]. Cardio-facio-cutaneous syndrome (CFC) is an autosomal dominant disease that occurs as a consequence of de novo heterozygous mutations in the BRAF, MAP2K1, MAP2K2 and KRAS genes. BRAF is the most frequently mutated gene, leading to a gain of function with consequent hyperactivation of the RAS/MAPK pathway [44]. Mutations affecting MAP2K1 and MAP2K2, known as MEK1 and MEK2, occur in the self-inhibitory region, leading to continuous activation of the kinases, which phosphorylate and activate ERK1 and ERK2 [45].
2.5. Inibition of RAS Downstream Targets
Based on these data, the inhibition of RAS downstream targets, such as MEK, has been proposed as a therapeutic option in RASopathies.
Several phase 2 clinical trials have been conducted using drugs that act on the Ras pathway and others such as pirfenidone (fibroblast inhibitor) [46], tipifarnib (farnesyl transferase inhibitor) [47][48], sirolimus (mTOR inhibitor) [49][50] but none of these has led to benefits that justified their use. A 20% reduction in PNF size has been observed with imatinib (a tyrosine kinase inhibitor) but only in 5% of patients [51] whereas pegylated interferon (a growth inhibitor, antiviral and immunomodulator agent) [52] had some effect only in 14% of patients [53]. Several MEK inhibitors have been tested in clinical trials in adults with refractory cancer. Solumetinib is a specific oral MEK 1/2 inhibitor and has been tested in Phase 1 clinical trials in children with plexiform neurofibromas (PNs) and low-grade gliomas [54]. In these studies, tumors were shown to shrink by 20% in 17 out of 24 children with side effects such as gastrointestinal upset, acneiform rush [55] but without serious toxic effects such as ocular toxicity and heart problems [56]. Phase 2 studies (SPRINT TRIAL) confirmed that 70% of patients treated with Solumetinib had a 20% reduction in pNF with a maximum response after 16 cycles [57]. On the basis of these results, Solumetinib received FDA approval for children 2 years of age and older with NF1 and inoperable and symptomatic PNs. Based on these encouraging data, other MEK inhibitors have been developed and tested in trials.
Trametinib, already approved for adult melanoma, is being tested in children with NF1-related PNs with size reduction by 20% in 46% for children with good tolerability [58]. Mirdametinib has similar results in 16-year-old patients with inoperable pNF45.
3. Ciliopathies
The term “ciliopathic” comes from the Bardet–Biedl syndrome (BBS). Later, it was observed that the malfunction of the cilia manifests itself with a great variety of characteristics common to different syndromes [59]. The term ciliopathy includes very different pathologies that share a common etiology and pathogenesis in mutations in genes involved in the functioning of the cilia. Cilia are scattered on the apical portion of epithelial cells throughout the body. They perform various functions by regulating the cell cycle, developing and maintaining cell polarity, and conducting mechanosensation. Cilia are divided into two groups, one represented by motile cilia and the other represented by non-motile cilia, also called primary cilia [60]. Motile cilia are found on respiratory epithelial cells, ependymal cells of the cerebrospinal fluid spaces, sperm cells, and embryonic node cells during development. Primary cilia act as an antenna for extracellular information and by converting mechanical or chemical stimuli into electrical signals [61]. For signal conversion, there are many Ca2+-permeable channels both on the membrane of the cilia and on the basal body, such as polycystin 2 (PC-2), and member 4 of the cation channel subfamily of the transient receptor potential V (TRPV4) (Figure 3 and Figure 4) [62].
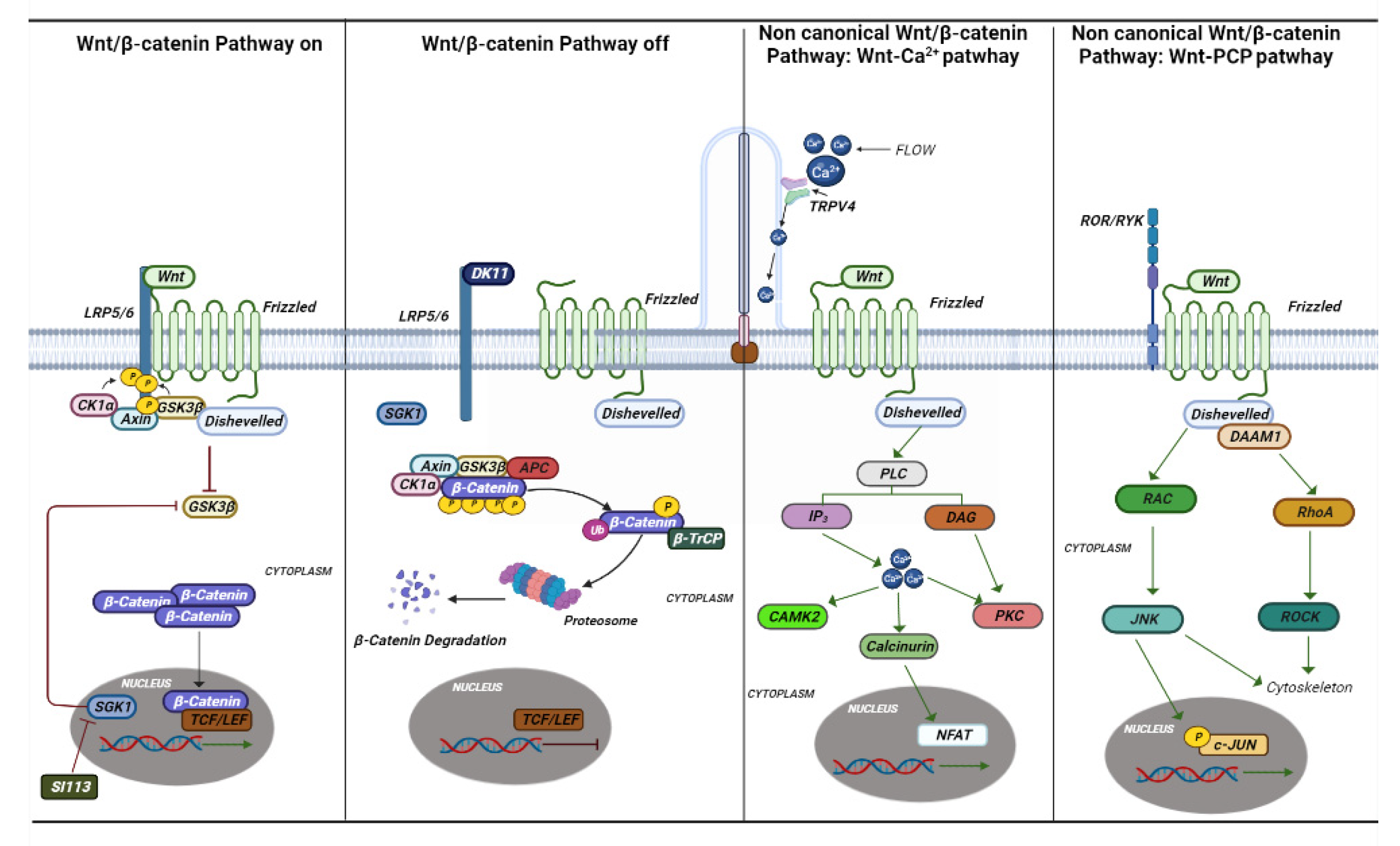
Figure 3. Canonical and non-canonical Wnt pathways. The canonical and noncanonical Wnt pathways are depicted in the figure with the various genes and mechanisms that are activated. In ciliopathies, several studies have shown that this pathway plays an essential role in their onset. Si113 is indicated; an SGK1 kinase inhibitor appears to have an effect on hydrocephalus caused by some ciliopathies.
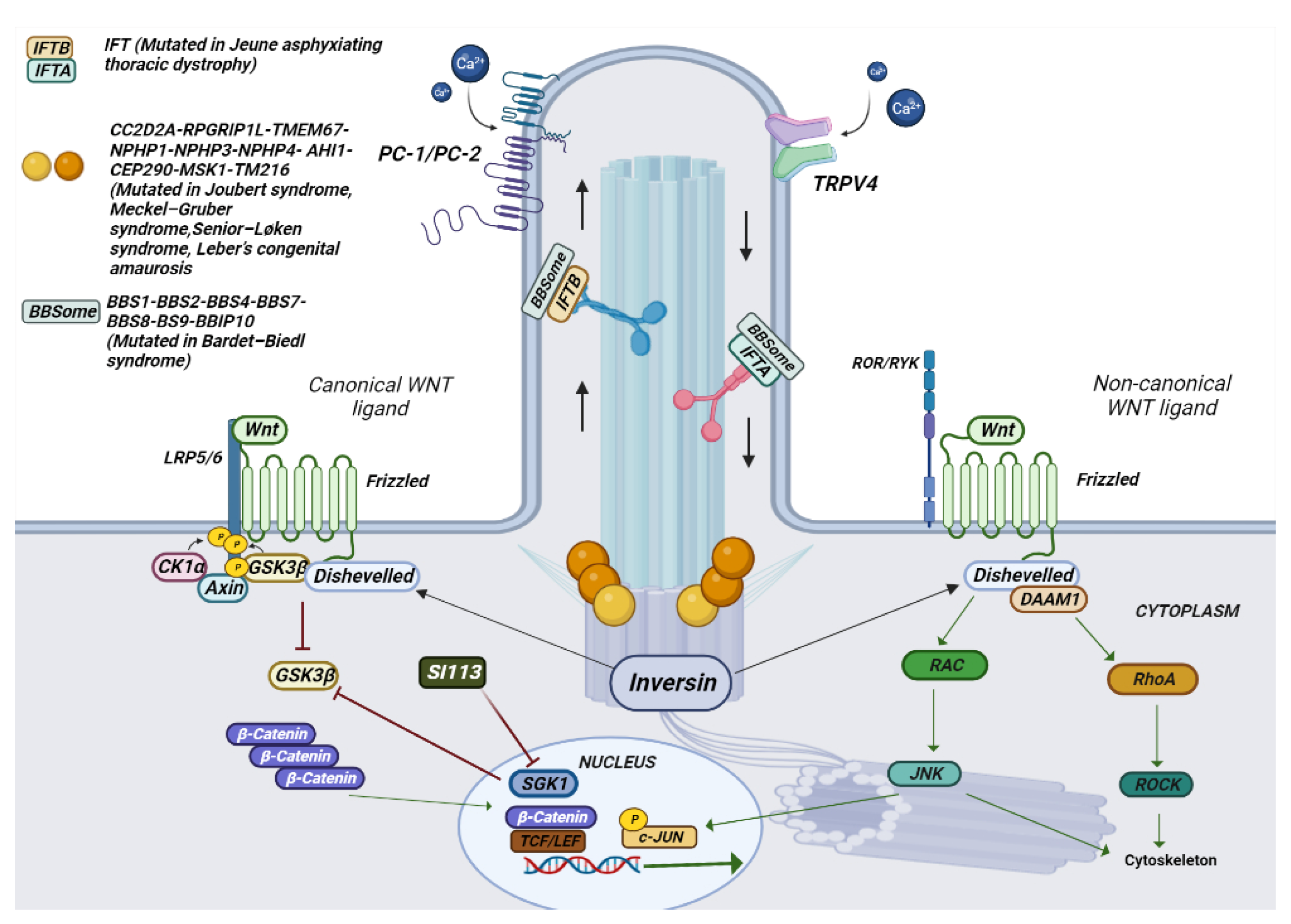
Figure 4. Schematic representation of the composition of a cilium and the various mutated genes in ciliopathies. In the photo it is possible to observe how a cilium is structured in its various components. The different genes mutated in ciliopathies, and their location are represented.
3.1. Clinical Manifestation
Joubert syndrome is a rare syndrome characterized by hypotonia, ataxia, psychomotor delay, irregular breathing pattern, and oculomotor apraxia [59].
Meckel–Gruber syndrome (MKS) is phenotypically similar to Joubert syndrome (JBTS) and has a characteristic phenotype with occipital encephalocele and other posterior fossa defects, cystic dysplastic kidneys, hepatic bile duct proliferation, and polydactyly. Senior–Løken syndrome leads to retinitis pigmentosa (RP) and renal disease and infantile onset, with several similarities to Joubert’s syndrome. Orofacial syndrome type 1 is a rare X-linked dominant disorder that leads to spontaneous abortions of male fetuses. Features include developmental defects of the oral cavity, face, and fingers;central nervous system (CNS) abnormalities; and cystic kidney disease [63]. Leber’s congenital amaurosis is characterized by retinal dystrophy which occurs during the first year. A characteristic symptom is Franceschetti’s oculodigital sign, consisting of pricking, pressing, and rubbing the eye [63]. Bardet–Biedl syndrome is characterized by renal malformations, with dysplasia nephrophtisis and cystic tubular disease, [64] polydactyl obesity, hypogonadism, and craniofacial defects [65]. Rhins syndrome is characterized by retinitis pigmentosa, hypopituitarism, nephronophthisis, and Skeletal dysplasia (RHYNS).
3.2. The Genetics of Ciliopathies
Mutations in the gene-encoding proteins involved in the development and signal conversion by cilia lead to developmental and degenerative diseases, which exhibit a broad phenotypic spectrum. The cilia perform their action by mediating key developmental pathways, such as Wnt and Shh signaling (Figure 3 and Figure 4). The binding of Wnt to its receptor complex composed of the Frizzled receptor (Fz) family and low-density lipoprotein-related protein 5/6 (LRP5/6) [66] activates various cascades of intracellular signal transduction, the signal activates disheveled (Dsh/Dvl), a cytoplasmic phosphoprotein, at the level of Dsh. This signal can take different routes; one of these is the canonical route (Wnt/β-catenin-dependent), and another is the non-canonical pathway (β-catenin-independent), which further subdivides into the planar cell polarity pathway and the Wnt/Ca2+ pathway (Figure 3) [67].
The canonical pathway of Wnt is characterized by the accumulation and translocation in the nucleus of a protein called β-catenin, which is very important in cell adhesion. When Wnt is not bound to its receptor, cytoplasmic β-catenin is phosphorylated by a complex of destruction consisting of Axin, adenomatosis polyposis coli (APC), protein phosphatase 2A (PP2A), glycogen synthase kinase 3 (GSK3), and casein kinase 1α (CK1α). Phosphorylated β-catenin is recognized for ubiquitination, resulting in proteasomal degradation. Binding of Wnt to its receptor complex induces membrane translocation of a negative regulator of Axin signaling, which binds to a conserved sequence in the cytoplasmic tail of LRP5/6 leading to disruption of the APC/Axin/GSK3 complex [66]. The stabilized β-catenin then translocates into the nucleus, where it enhances the transcription of genes regulating several cellular functions related to the development of a neoplastic phenotype, i.e., inflammation, fibrosis, Warburg effect and metabolism, and circadian rhythms. The transcriptional output of beta catenin is mediated by cyclin-dependent kinases, such as cyclin-dependent kinase 8 (CDK8), 19 module [68][69].
TMEM67, one of a complex of proteins involved in formation of the primary cilium, may be considered a prototype gene coding for a protein that inhibits canonical Wnt through the repression of the expression of homeoboxtype transcription factors, including the HOX5 gene group [70]. Consistent with a role in non-canonical Wnt signalling, TMEM67 is required for the migration of centrioli towards the apical membrane and for the regulation of actin cytoskeleton remodeling. TMEM67 (TMEM67−/−) knock-out animals recapitulate several features of MKS, Joubert syndrome, and Rhyns syndrome in humans and correlate with an aberrant increase in Wnt/β-catenin signaling both in vitro and in vivo [71][72].
3.3. Inibition of TRPV4 and SGK1
In a porcine CP epithelial cell line, stimulation of endogenous TRPV4 results in multiphasic ion fluxes, which can be blocked by either of two specific TRPV4 antagonists [73]. Interestingly, in a unique rat model MKS, the Tmem67 genetic model [74], treatment with TRPV4 antagonists (HC067047 and RN 1734) counteracted the development of the hydrocephalic phenotype in homozygous TMEM67−/− rats. On the other hand, TRPV4 agonist treatment exacerbated the hydrocephalus in the affected animals [75]. Given the peculiar role that Sgk1 seems to maintain in modulating canonical WNT signaling and in channel regulation, it is possible that Sgk1 inhibition can also have a role in reducing some of the phenotypic characteristics of ciliopathyes characterized by a dysregulated Wnt canonical pathway (Figure 4).
References
- Osborne, J.P.; Fryer, A.; Webb, D. Epidemiology of Tuberous Sclerosis. Ann. New York Acad. Sci. 1991, 615, 125–127.
- Curatolo, P.; Bombardieri, R.; Jozwiak, S. Tuberous sclerosis. Lancet 2008, 372, 657–668.
- DiMario, F.J. Brain Abnormalities in Tuberous Sclerosis Complex. J. Child Neurol. 2004, 19, 650–657.
- Luat, A.F.; Makki, M.; Chugani, H.T. Neuroimaging in tuberous sclerosis complex. Curr. Opin. Neurol. 2007, 20, 142–150.
- Islam, M.P.; Roach, E.S. Tuberous sclerosis complex. Handb. Clin. Neurol. 2015, 132, 97–109.
- Kaczorowska, M.; Jurkiewicz, E.; Domańska-Pakieła, D.; Syczewska, M.; Lojszczyk, B.; Chmielewski, D.; Kotulska, K.; Kuczyński, D.; Kmieć, T.; Dunin-Wąsowicz, D.; et al. Cerebral tuber count and its impact on mental outcome of patients with tuberous sclerosis complex. Epilepsia 2011, 52, 22–27.
- Capal, J.K.; Bernardino-Cuesta, B.; Horn, P.S.; Murray, D.; Byars, A.W.; Bing, N.M.; Kent, B.; Pearson, D.A.; Sahin, M.; Krueger, D.A. Influence of seizures on early development in tuberous sclerosis complex. Epilepsy Behav. 2017, 70, 245–252.
- Jansen, F.E.; Vincken, K.L.; Algra, A.; Anbeek, P.; Braams, O.; Nellist, M.; Zonnenberg, B.A.; Jennekens-Schinkel, A.; Ouweland, A.V.D.; Halley, D.; et al. Cognitive impairment in tuberous sclerosis complex is a multifactorial condition. Neurology 2007, 70, 916–923.
- Ehninger, D.; Sano, Y.; De Vries, P.J.; Dies, K.; Franz, D.; Geschwind, D.H.; Kaur, M.; Lee, Y.-S.; Li, W.; Lowe, J.K.; et al. Gestational immune activation and Tsc2 haploinsufficiency cooperate to disrupt fetal survival and may perturb social behavior in adult mice. Mol. Psychiatry 2010, 17, 62–70.
- Wu, S.; Wu, F.; Jiang, Z. Identification of hub genes, key miRNAs and potential molecular mechanisms of colorectal cancer. Oncol. Rep. 2017, 38, 2043–2050.
- Van Slegtenhorst, M.; de Hoogt, R.; Hermans, C.; Nellist, M.; Janssen, B.; Verhoef, S.; Lindhout, D.; van den Ouweland, A.; Halley, D.; Young, J.; et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997, 277, 805–808.
- Sancak, O.; Nellist, M.; Goedbloed, M.; Elfferich, P.; Wouters, C.; Maat-Kievit, A.; Zonnenberg, B.; Verhoef, S.; Halley, D.; Ouweland, A.V.D. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: Genotype—Phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur. J. Hum. Genet. 2005, 13, 731–741.
- Cheadle, J.P.; Reeve, M.P.; Sampson, J.R.; Kwiatkowski, D.J. Molecular genetic advances in tuberous sclerosis. Hum. Genet. 2000, 107, 97–114.
- Kwiatkowski, D.J.; Manning, B.D. Molecular Basis of Giant Cells in Tuberous Sclerosis Complex. N. Engl. J. Med. 2014, 371, 778–780.
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555.
- Tee, A.R.; Fingar, D.C.; Manning, B.D.; Kwiatkowski, D.J.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13571–13576.
- Crino, P.B. mTOR: A pathogenic signaling pathway in developmental brain malformations. Trends Mol. Med. 2011, 17, 734–742.
- Crino, P.B.; Nathanson, K.L.; Henske, E.P. The tuberous sclerosis complex. N. Engl. J. Med. 2006, 355, 1345–1356.
- Huang, J.; Manning, B.D. The TSC1–TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190.
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193.
- Carson, R.P.; Fu, C.; Winzenburger, P.; Ess, K.C. Deletion of Rictor in neural progenitor cells reveals contributions of mTORC2 signaling to tuberous sclerosis complex. Hum. Mol. Genet. 2012, 22, 140–152.
- Vézina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. 1975, 28, 721–726.
- Franz, D.N.; Krueger, D.A. mTOR inhibitor therapy as a disease modifying therapy for tuberous sclerosis complex. Am. J. Med. Genet. Part C Semin. Med. Genet. 2018, 178, 365–373.
- Krueger, D.A.; Care, M.M.; Holland, K.; Agricola, K.; Tudor, C.; Mangeshkar, P.; Wilson, K.A.; Byars, A.; Sahmoud, T.; Franz, D.N. Everolimus for Subependymal Giant-Cell Astrocytomas in Tuberous Sclerosis. N. Engl. J. Med. 2010, 363, 1801–1811.
- Franz, D.N.; Belousova, E.; Sparagana, S.; Bebin, E.M.; Frost, M.; Kuperman, R.; Witt, O.; Kohrman, M.H.; Flamini, J.R.; Wu, J.Y.; et al. Everolimus for subependymal giant cell astrocytoma in patients with tuberous sclerosis complex: 2-year open-label extension of the randomised EXIST-1 study. Lancet Oncol. 2014, 15, 1513–1520.
- Rauen, K.A. The RASopathies. Annu. Rev. Genom. Hum. Genet. 2013, 14, 355–369.
- Bergqvist, C.; Network, N.F.; Servy, A.; Valeyrie-Allanore, L.; Ferkal, S.; Combemale, P.; Wolkenstein, P. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J. Rare Dis. 2020, 15, 37.
- Darrigo, L.G.; Geller, M.; Bonalumi Filho, A.; Azulay, D.R. Prevalence of plexiform neurofibroma in children and ado-lescents with type I neurofibromatosis. J. Pediatr. (Rio J.) 2007, 83, 571–573.
- Gross, A.M.; Singh, G.; Akshintala, S.; Baldwin, A.; Dombi, E.; Ukwuani, S.; Goodwin, A.; Liewehr, D.J.; Steinberg, S.M.; Widemann, B.C. Association of plexiform neurofibroma volume changes and development of clinical morbidities in neurofibromatosis 1. Neuro-Oncology 2018, 20, 1643–1651.
- Nguyen, R.; Kluwe, L.; Fuensterer, C.; Kentsch, M.; Friedrich, R.E.; Mautner, V.-F. Plexiform neurofibromas in children with neurofibromatosis type 1: Frequency and associated clinical deficits. J. Pediatr. 2011, 159, 652–655.e2.
- Weiss, B.; Bollag, G.; Shannon, K. Hyperactive Ras as a therapeutic target in neurofibromatosis type 1. Am. J. Med. Genet. 1999, 89, 14–22.
- Tartaglia, M.; Zampino, G.; Gelb, B. Noonan Syndrome: Clinical Aspects and Molecular Pathogenesis. Mol. Syndr. 2010, 1, 2–26.
- Tartaglia, M.; Mehler, E.L.; Goldberg, R.; Zampino, G.; Brunner, H.G.; Kremer, H.; Van Der Burgt, I.; Crosby, A.H.; Ion, A.; Jeffery, S.; et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 2001, 29, 465–468.
- Matozaki, T.; Murata, Y.; Saito, Y.; Okazawa, H.; Ohnishi, H. Protein tyrosine phosphatase SHP-2: A proto-oncogene product that promotes Ras activation. Cancer Sci. 2009, 100, 1786–1793.
- Quilliam, L.A.; Rebhun, J.F.; Castro, A.F. A growing family of guanine nucleotide exchange factors is responsible for activation of ras-family GTPases. In Progress in Nucleic Acid Research and Molecular Biology; Academic Press: Cambridge, MA, USA, 2002; Volume 71, pp. 391–444.
- Tartaglia, M.; Pennacchio, L.; Zhao, C.; Yadav, K.K.; Fodale, V.; Sarkozy, A.; Pandit, B.; Oishi, K.; Martinelli, S.; Schackwitz, W.; et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat. Genet. 2006, 39, 75–79.
- Gremer, L.; Merbitz-Zahradnik, T.; Dvorsky, R.; Cirstea, I.C.; Kratz, C.P.; Zenker, M.; Wittinghofer, A.; Ahmadian, M.R. Germline KRAS mutations cause aberrant biochemical and physical properties leading to developmental disorders. Hum. Mutat. 2010, 32, 33–43.
- Schubbert, S.; Bollag, G.; Lyubynska, N.; Nguyen, H.; Kratz, C.P.; Zenker, M.; Niemeyer, C.M.; Molven, A.; Shannon, K. Bi-ochemical and Functional Characterization of Germ Line KRAS Mutations. Mol. Cell. Biol. 2007, 27, 7765–7770.
- Pandit, B.; Sarkozy, A.; Pennacchio, L.A.; Carta, C.; Oishi, K.; Martinelli, S.; Pogna, E.A.; Schackwitz, W.; Ustaszewska, A.; Landstrom, A.; et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyo-pathy. Nat. Genet. 2007, 39, 1007–1012.
- Mutation Analysis of the SHOC2 Gene in Noonan-Like Syndrome and in Hematologic Malignancies|Journal of Human Ge-netics. Available online: https://www.nature.com/articles/jhg2010116 (accessed on 13 January 2023).
- Martinelli, S.; De Luca, A.; Stellacci, E.; Rossi, C.; Checquolo, S.; Lepri, F.; Caputo, V.; Silvano, M.; Buscherini, F.; Consoli, F.; et al. Heterozygous Germline Mutations in the CBL Tumor-Suppressor Gene Cause a Noonan Syndrome-like Phenotype. Am. J. Hum. Genet. 2010, 87, 250–257.
- Gripp, K.W.; Morse, L.A.; Axelrad, M.; Chatfield, K.C.; Chidekel, A.; Dobyns, W.; Doyle, D.; Kerr, B.; Lin, A.E.; Schwartz, D.D.; et al. Costello syn-drome: Clinical phenotype, genotype, and management guidelines. Am. J. Med. Genet. Part A 2019, 179, 1725–1744.
- Zenker, M. Noonan Syndrome and Related Disorders: A Matter of Deregulated Ras Signaling; Karger Medical and Scientific Publishers: Basel, Switzerland, 2009.
- Nava, C.; Hanna, N.; Michot, C.; Pereira, S.; Pouvreau, N.; Niihori, T.; Aoki, Y.; Matsubara, Y.; Arveiler, B.; Lacombe, D.; et al. Cardio-facio-cutaneous and Noonan syndromes due to mutations in the RAS/MAPK signalling pathway: Genotype phenotype relationships and overlap with Costello syndrome. J. Med. Genet. 2007, 44, 763–771.
- Ordan, M.; Pallara, C.; Maik-Rachline, G.; Hanoch, T.; Gervasio, F.L.; Glaser, F.; Fernandez-Recio, J.; Seger, R. Intrinsically active MEK variants are differentially regulated by proteinases and phosphatases. Sci. Rep. 2018, 8, 11830.
- Widemann, B.C.; Babovic-Vuksanovic, D.; Dombi, E.; Wolters, P.L.; Goldman, S.; Martin, S.; Goodwin, A.; Goodspeed, W.; Kieran, M.W.; Cohen, B.; et al. Phase II trial of pirfenidone in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Pediatr. Blood Cancer 2014, 61, 1598–1602.
- Widemann, B.C.; Salzer, W.L.; Arceci, R.J.; Blaney, S.M.; Fox, E.; End, D.; Gillespie, A.; Whitcomb, P.; Palumbo, J.S.; Pitney, A.; et al. Phase I Trial and Pharmacokinetic Study of the Farnesyltransferase Inhibitor Tipifarnib in Children With Refractory Solid Tumors or Neurofibromatosis Type I and Plexiform Neurofibromas. J. Clin. Oncol. 2006, 24, 507–516.
- Widemann, B.C.; Dombi, E.; Gillespie, A.; Wolters, P.L.; Belasco, J.; Goldman, S.; Korf, B.R.; Solomon, J.; Martin, S.; Salzer, W.; et al. Phase 2 ran-domized, flexible crossover, double-blinded, placebo-controlled trial of the farnesyltransferase inhibitor tipi-farnib in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Neuro Oncol. 2014, 16, 707–718.
- Weiss, B.; Widemann, B.C.; Wolters, P.; Dombi, E.; Vinks, A.; Cantor, A.; Perentesis, J.; Schorry, E.; Ullrich, N.; Gutmann, D.H.; et al. Sirolimus for progressive neurofibromatosis type 1-associated plexiform neurofibromas: A Neurofibromatosis Clinical Trials Consortium phase II study. Neuro-Oncology 2014, 17, 596–603.
- Weiss, B.; Widemann, B.C.; Wolters, P.; Dombi, E.; Vinks, A.A.; Cantor, A.; Korf, B.; Perentesis, J.; Gutmann, D.H.; Schorry, E.; et al. Sirolimus for non-progressive NF1-associated plexiform neurofibromas: An NF clinical trials consortium phase II study. Pediatr. Blood Cancer 2013, 61, 982–986.
- Jakacki, R.I.; Dombi, E.; Potter, D.M.; Goldman, S.; Allen, J.C.; Pollack, I.F.; Widemann, B.C. Phase I trial of pegylated interferon- -2b in young patients with plexiform neurofibromas. Neurology 2011, 76, 265–272.
- Jakacki, R.I.; Dombi, E.; Steinberg, S.M.; Goldman, S.; Kieran, M.W.; Ullrich, N.J.; Pollack, I.F.; Goodwin, A.; Manley, P.E.; Fangusaro, J.; et al. Phase II trial of pegylated interferon alfa-2b in young patients with neu-rofibromatosis type 1 and unresectable plexiform neurofibromas. Neuro Oncol. 2017, 19, 289–297.
- Robertson, K.A.; Nalepa, G.; Yang, F.-C.; Bowers, D.C.; Ho, C.Y.; Hutchins, G.D.; Croop, J.M.; Vik, T.A.; Denne, S.C.; Parada, L.F.; et al. Imatinib mesylate for plexiform neurofibromas in patients with neurofibromatosis type 1: A phase 2 trial. Lancet Oncol. 2012, 13, 1218–1224.
- Banerjee, A.; Jakacki, R.I.; Onar-Thomas, A.; Wu, S.; Nicolaides, T.; Poussaint, T.Y.; Fangusaro, J.; Phillips, J.; Perry, A.; Turner, D.; et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: A Pediatric Brain Tumor Consortium (PBTC) study. Neuro-Oncology 2017, 19, 1135–1144.
- Dombi, E.; Baldwin, A.; Marcus, L.J.; Fisher, M.J.; Weiss, B.; Kim, A.; Whitcomb, P.; Martin, S.; Aschbacher-Smith, L.E.; Rizvi, T.A.; et al. Activity of Selumetinib in Neurofibromatosis Type 1–Related Plexiform Neurofibromas. N. Engl. J. Med. 2016, 375, 2550–2560.
- Gross, A.; Bishop, R.; Widemann, B.C. Selumetinib in Plexiform Neurofibromas. N. Engl. J. Med. 2017, 376, 1195.
- Plotkin, S.R.; Blakeley, J.O.; Dombi, E.; Fisher, M.J.; Hanemann, C.O.; Walsh, K.; Wolters, P.L.; Widemann, B.C. Achieving consensus for clinical trials: The REiNS International Collaboration. Neurology 2013, 81, S1–S5.
- McCowage, G.B.; Mueller, S.; Pratilas, C.A.; Hargrave, D.R.; Moertel, C.L.; Whitlock, J.; Fox, E.; Hingorani, P.; Russo, M.W.; Dasgupta, K.; et al. Trametinib in pediatric patients with neurofibromatosis type 1 (NF-1)–associated plexiform neurofibroma: A phase I/IIa study. J. Clin. Oncol. 2018, 36, 10504.
- Waters, A.M.; Beales, P.L. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056.
- Oud, M.M.; Lamers, I.J.C.; Arts, H.H. Ciliopathies: Genetics in Pediatric Medicine. J. Pediatr. Genet. 2016, 6, 018–029.
- Khayyeri, H.; Barreto, S.; Lacroix, D. Primary cilia mechanics affects cell mechanosensation: A computational study. J. Theor. Biol. 2015, 379, 38–46.
- Sonkusare, S.K.; Bonev, A.D.; Ledoux, J.; Liedtke, W.; Kotlikoff, M.I.; Heppner, T.J.; Hill-Eubanks, D.C.; Nelson, M.T. Elementary Ca2+ Signals Through Endothelial TRPV4 Channels Regulate Vascular Function. Science 2012, 336, 597–601.
- Feather, S.A.; Winyard, P.J.; Dodd, S.; Woolf, A. Oral-facial-digital syndrome type 1 is another dominant polycystic kidney disease: Clinical, radiological and histopathological features of a new kindred. Nephrol. Dial. Transplant. 1997, 12, 1354–1361.
- Beales, P.L.; Elcioglu, N.; Woolf, A.S.; Parker, D.; Flinter, F.A. New criteria for improved diagnosis of Bardet-Biedl syn-drome: Results of a population survey. J. Med. Genet. 1999, 36, 437–446.
- Tobin, J.L.; Di Franco, M.; Eichers, E.; May-Simera, H.; Garcia, M.; Yan, J.; Quinlan, R.; Justice, M.J.; Hennekam, R.C.; Briscoe, J.; et al. Inhibition of neural crest migration underlies craniofacial dysmorphology and Hirschsprung’s disease in Bardet–Biedl syndrome. Proc. Natl. Acad. Sci. USA 2008, 105, 6714–6719.
- He, X.; Semenov, M.; Tamai, K.; Zeng, X. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: Arrows point the way. Development 2004, 131, 1663–1677.
- Habas, R.; Dawid, I.B. Dishevelled and Wnt signaling: Is the nucleus the final frontier? J. Biol. 2005, 4, 2.
- LeCarpentier, Y.; Schussler, O.; Hébert, J.-L.; Vallée, A. Multiple Targets of the Canonical WNT/β-Catenin Signaling in Cancers. Front. Oncol. 2019, 9, 1248.
- Bian, J.; Dannappel, M.; Wan, C.; Firestein, R. Transcriptional Regulation of Wnt/β-Catenin Pathway in Colorectal Cancer. Cells 2020, 9, 2125.
- Abdelhamed, Z.A.; Abdelmottaleb, D.I.; El-Asrag, M.E.; Natarajan, S.; Wheway, G.; Inglehearn, C.F.; Toomes, C.; Johnson, C.A. The ciliary Frizzled-like receptor Tmem67 regulates canonical Wnt/β-catenin signalling in the developing cerebellum via Hoxb5. Sci. Rep. 2019, 9, 5446.
- Brancati, F.; Italy, U.D.N.; Camerota, L.; Colao, E.; Vega-Warner, V.; Zhao, X.; Zhang, R.; Bottillo, I.; Castori, M.; Caglioti, A.; et al. Biallelic variants in the ciliary gene TMEM67 cause RHYNS syndrome. Eur. J. Hum. Genet. 2018, 26, 1266–1271.
- Abdelhamed, Z.A.; Wheway, G.; Szymanska, K.; Natarajan, S.; Toomes, C.; Inglehearn, C.; Johnson, C.A. Variable expressivity of ciliopathy neurological phenotypes that encompass Meckel-Gruber syn-drome and Joubert syndrome is caused by complex de-regulated ciliogenesis, Shh and Wnt signalling defects. Hum. Mol. Genet. 2013, 22, 1358–1372.
- Bagher, P.; Beleznai, T.; Kansui, Y.; Mitchell, R.; Garland, C.J.; Dora, K.A. Low intravascular pressure activates endothelial cell TRPV4 channels, local Ca 2+ events, and IK Ca channels, reducing arteriolar tone. Proc. Natl. Acad. Sci. USA 2012, 109, 18174–18179.
- Preston, D.; Simpson, S.; Halm, D.; Hochstetler, A.; Schwerk, C.; Schroten, H.; Blazer-Yost, B.L. Activation of TRPV4 stimulates transepithelial ion flux in a porcine choroid plexus cell line. Am. J. Physiol. Physiol. 2018, 315, C357–C366.
- Hochstetler, A.E.; Smith, H.M.; Preston, D.C.; Reed, M.M.; Territo, P.R.; Shim, J.W.; Fulkerson, D.; Blazer-Yost, B.L. TRPV4 antagonists ameliorate ventriculomegaly in a rat model of hydrocephalus. JCI Insight 2020, 5, 137646.
More
Information
Subjects:
Others
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
749
Revisions:
2 times
(View History)
Update Date:
17 Mar 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No