Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Nicola Perrotti and Version 2 by Lindsay Dong.
Gene variants may have functional consequences on the protein product. The variants can be classified as loss of function (LOF) when the protein function is reduced or lost and gain of function (GOF) when the protein function is enhanced or a new function is acquired. Kinase inhibitors have been used for many years in tumor therapy, and new monoclonal antibodies—as well as small inhibitory molecules with different indications in tumor therapy—are approved every year by the regulatory agencies. human epidermal growth factor receptor 2 (HER2) inhibitors are currently used in HER2-positive carcinomas, including breast, colon, and non-small-cell lung (NSCLC) cancers, Abl inhibitors are used in chronic myeloid leukemia.
- kinase inhibitors
- genetics
- RAS pathway
- mTOR pathway
- Wnt pathway
1. Tuberous Sclerosis Complex
TSC is a disease characterized by autosomal dominant inheritance and has an incidence of 1 in 6000–10,000 individuals [1][22]. It is involved in the development of very different pathologies that involve the brain, heart, kidneys, and skin.
1.1. Clinical Manifestation
Neurological manifestations represent the most important cause of impairment in most patients. These include epilepsy, present in 90% of patients [2][23]; cortical tubers; subependymal nodules; giant cell astrocytomas; intellectual disability; autistic spectrum disorder; and behavioral problems [3][4][24,25].
Most patients with epilepsy have seizures in the first year of age [5][26]. Some studies have speculated that the seizures originate from cortical tubers, but the mechanism is still being studied [6][27]. Long-term damage depends on the age of onset and the severity of seizures [7][8][28,29].
The cortical tuber, from which the disease takes its name, is a focal malformation present in 80–90% of patients. It is due to a failure of cell differentiation with neuronal migration during neurological development [9][30].
1.2. Genetics of TSC
TSC is caused by mutations in two genes, the TSC1 gene located on chromosome 9 (9q34), which encodes the hamartina protein, and TSC2 located on chromosome 16 (16p13.3), which encodes the tuberin protein [10][11][37,38]. TSC1 mutations have been identified in ~10–20% of patients, whereas TSC2 mutations have been identified in ~70–90% of patients clinically diagnosed with TSC [12][13][39,40]. The proteins produced by these two genes physically interact with high affinity to form a heterotrimeric complex, termed the TSC protein complex, with the TBC1 domain family member 7 (TBC1D7), [14][15][41,42] and act on the signaling pathway of mTORC1 (rapamycin complex 1), a serine/threonine kinase involved in many cellular processes, such as cell growth, proliferation, and response to extracellular stress [16][17][18][43,44,45]. The major driver of the cellular hyperplasia and tissue dysplasia seen in TSC is the overactivation of the mTORC1 signaling pathway. Under normal conditions, growth factors stimulate the PI3K (phosphatidylinositol 3-kinase) and Ras–MAPK (mitogen-activated protein kinase) pathways, thereby inhibiting the TSC protein complex and activating mTOR signaling (Figure 1) [19][20][46,47]. mTOR acts through two complexes, mTORC1 and mTORC2. The loss of function of TSC genes results in an increase in mTORC1 and a decrease in mTORC2 signaling [21][48]. The mechanism by which this occurs is not clear; it is probably due to loss of direct binding of the amartin/tuberin complex to mTORC2. Alternatively, increasing mTORC1 activates a negative feedback loop from p70S6K inhibiting IRS-1 by inhibiting the PI3-kinase-dependent activation of mTORC2.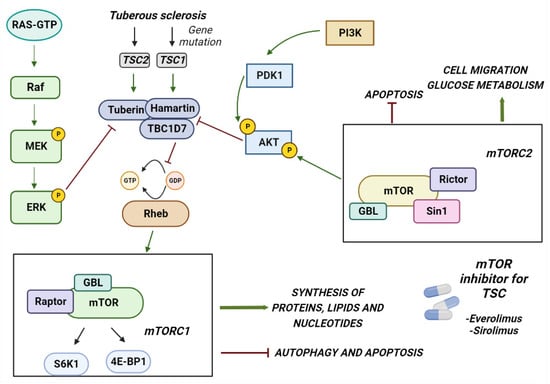
Figure 1. The figure shows how the mTORC1 pathway is involved in the development of TSC. The figure shows the genes involved in the development of TSC and the pathway activated by different proteins. In particular, mTORC1 may contribute to the symptoms of TSC when hyperactivated by the malfunction of the TBC1D7 tuberin and hamartin complex. Two of the FDA-approved kinase inhibitors for the treatment of TSC are also shown in the figure.
1.3. Inhibition of mTOR
The mTOR inhibitors in use today are sirolimus and everolimus. The former is also known as rapamycin and has been obtained from soil samples from Easter Island [22][58]. Everolimus is a sirolimus derivative obtained via the addition of an ethyl ester group. Both compounds induce allosteric dissociation of the cofactor rapTOR (TOR regulation-related protein) that is essential for mTOR function [23][59]. TSC-associated epilepsy does not respond well to treatment with traditional antiepileptics. Some trials show that mTOR inhibition can reduce the frequency of epileptic manifestations and reduce the volume of tumors and nodules [24][25][60,61].
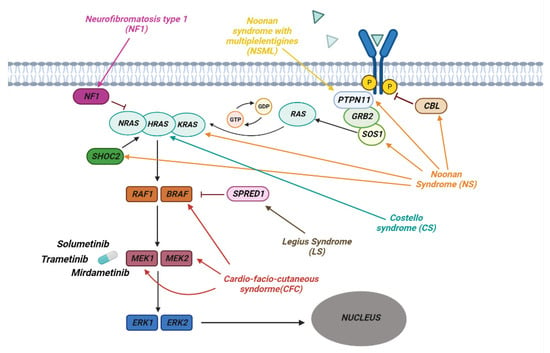
2. RASopathies
RASopathies affect about 1 in 1000 individuals and are part of a heterogeneous group of diseases in which there is a germline mutation in the genes that code for components and regulators of the Ras/MAPK Pathway (Figure 2). This is one of the best-studied and characterized pathways and is involved in the regulation of growth, cell cycle differentiation, and normal mammalian development.Figure 2. The pathway of RAS/MAPK in RASopathies. The figure shows how the various genes of the Ras/MAPK pathway are involved in the development of the different syndromes associated with RASopathies. Shown are some of the MERK1/2 kinase inhibitors used to treat RASopathies, some of which are FDA-approved.
2.1. Clinical Manifestation
These pathologies each have their own peculiar phenotype but have common characteristics, such as cardiac malformations, craniofacial dysmorphologies, skin, ocular and musculoskeletal abnormalities, hypotonia, and predisposition to tumors [26][75].2.2. Neurofibromatosis
The first to be identified was Neurofibromatosis type 1 (NF1). This is an autosomal dominant disease that affects 1 in 3000 individuals and is present in one of the parents in 50% of cases, whereas it is the result of a de novo mutation in the remaining 50%. NF1 is characterized by the development of progressive benign and malignant tumors of the central and peripheral nervous system [27][76]. Diagnosis is mainly based on the presence of café-au-lait maculae, intertriginous freckling, neurofibromas and plexiform neurofibromas, iris Lisch nodules, osseous dysplasia, optic pathway glioma, and other symptoms common to RASophaties in general. The most severe and frequent clinical manifestations in type 1 neurofibromatosis are plexiform neurofibromas (PNFs), low-grade gliomas, and other peripheral nerve sheath tumors. PNFs occurs in 20–50% of patients with NF1 [28][77]. These are tumors of the peripheral nerves that are generally benign but can cause complications such as disfigurement and pain [29][30][78,79]. PNFs are diagnosed in early childhood and may need to undergo surgical excision, although the procedure can be complicated by factors such as proximity to nerves and wide vascularity.2.3. Noonan Syndrome
Noonan syndrome (NS) is one of the most common among RASopathies, in most cases it is inherited in an autosomal dominant fashion, with a variable expression.74,75 Clinical diagnosis is based on peculiar facial characteristics, short stature, cardiac defects—such as pulmonary stenosis or hypetrophic cardiomyopathy—and variable degrees of development delay. Noonan syndrome clinically overlaps with other RASopathies that are the consequence of genetic defects in the same RAS pathway. In Noonan syndrome with multiple lentigines (NSML)—formerly known as Leopard syndrome, Costello Syndrome (CS), and Cardio-facio-cutaneous syndrome (CFC)—most of the features of Noonan syndrome are associated with severe mental retardation, severe feeding difficulties, myopathy, facial papillomata, and warts, particularly in the nasolabial area, loose skin on the hands and feet, with hyperkeratotic palms and soles.2.4. Genetics of RASopathies
On the basis of the mutated gene of the Ras/MAPK pathway, RASopathies are classified into different groups. Some syndromes are due to the mutation of a single gene in this pathway, while others can arise as a result of the mutation of several genes. NF1 is caused by a mutation in the NF1 gene encoding the neurofibromin which is RasGAP, a GTPase-activating protein, that regulates Ras negatively. This mutation results in a loss of protein function followed by increased Ras activity that explains the predisposition to the development of malignant tumors observed in NF1 patients [31][80]. Noonan Syndrome is due to germline pathogenic variants involving genes such as PTPN11, SOS1, KRAS, NRAS, SHOC2, CBL and MAP2K1 [32][81]. The most frequent mutations are in the PTPN11, SOS1, and KRAS genes. These are generally de novo missense mutations that, once established in the germline, can be inherited in an autosomal dominant fashion. It is generally believed that missense mutations in the PTPN11 gene coding for the SHP-2 tyrosine phosphatase cause a gain of function in the enzymatic activity [33][82] and that active SHP-2 positively regulates cell growth and differentiation by promoting activation of the Ras–MAPK pathway (Figure 2) [34][83]. Mutations involving the SOS1 gene, a member of the RAS GEF family important in the conversion of GDP into GTP, lead to the activation of RAS [35][36][84,85]. The most frequent mutations of this gene occur in the codons that code for the genes involved in the maintenance of its self-inhibited form, leading to an increase in the activity of SOS1 and a continuous activation of RAS [36][85]. Other frequent mutations that lead to the onset of this syndrome are those affecting KRAS, which lead to a reduction in the hydrolysis of GTP with accumulation of the active form of KRAS [37][38][86,87]. Other rarer mutations, leading to the development of Noonan Syndrome, have been observed in the BRAF, SHOC2, and CBL genes [39][40][41][88,89,90]. CS is an autosomal dominant disease characterized by germline mutations of the HRAS gene, located on chromosome 11p15.5. One of the most common variants is pGly12Ser, which leads to greater activation of HRAS following a reduction in GAP-induced GTPase activity [42][43][92,93]. Cardio-facio-cutaneous syndrome (CFC) is an autosomal dominant disease that occurs as a consequence of de novo heterozygous mutations in the BRAF, MAP2K1, MAP2K2 and KRAS genes. BRAF is the most frequently mutated gene, leading to a gain of function with consequent hyperactivation of the RAS/MAPK pathway [44][94]. Mutations affecting MAP2K1 and MAP2K2, known as MEK1 and MEK2, occur in the self-inhibitory region, leading to continuous activation of the kinases, which phosphorylate and activate ERK1 and ERK2 [45][95].2.5. Inibition of RAS Downstream Targets
Based on these data, the inhibition of RAS downstream targets, such as MEK, has been proposed as a therapeutic option in RASopathies. Several phase 2 clinical trials have been conducted using drugs that act on the Ras pathway and others such as pirfenidone (fibroblast inhibitor) [46][103], tipifarnib (farnesyl transferase inhibitor) [47][48][104,105], sirolimus (mTOR inhibitor) [49][50][106,107] but none of these has led to benefits that justified their use. A 20% reduction in PNF size has been observed with imatinib (a tyrosine kinase inhibitor) but only in 5% of patients [51][108] whereas pegylated interferon (a growth inhibitor, antiviral and immunomodulator agent) [52][109] had some effect only in 14% of patients [53][110]. Several MEK inhibitors have been tested in clinical trials in adults with refractory cancer. Solumetinib is a specific oral MEK 1/2 inhibitor and has been tested in Phase 1 clinical trials in children with plexiform neurofibromas (PNs) and low-grade gliomas [54][111]. In these studies, tumors were shown to shrink by 20% in 17 out of 24 children with side effects such as gastrointestinal upset, acneiform rush [55][112] but without serious toxic effects such as ocular toxicity and heart problems [56][113]. Phase 2 studies (SPRINT TRIAL) confirmed that 70% of patients treated with Solumetinib had a 20% reduction in pNF with a maximum response after 16 cycles [57][114]. On the basis of these results, Solumetinib received FDA approval for children 2 years of age and older with NF1 and inoperable and symptomatic PNs. Based on these encouraging data, other MEK inhibitors have been developed and tested in trials. Trametinib, already approved for adult melanoma, is being tested in children with NF1-related PNs with size reduction by 20% in 46% for children with good tolerability [58][115]. Mirdametinib has similar results in 16-year-old patients with inoperable pNF45.3. Ciliopathies
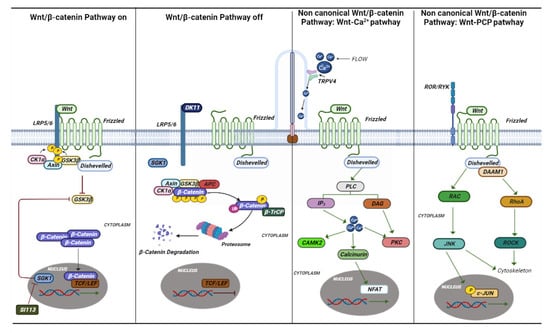
The term “ciliopathic” comes from the Bardet–Biedl syndrome (BBS). Later, it was observed that the malfunction of the cilia manifests itself with a great variety of characteristics common to different syndromes [59][125]. The term ciliopathy includes very different pathologies that share a common etiology and pathogenesis in mutations in genes involved in the functioning of the cilia. Cilia are scattered on the apical portion of epithelial cells throughout the body. They perform various functions by regulating the cell cycle, developing and maintaining cell polarity, and conducting mechanosensation. Cilia are divided into two groups, one represented by motile cilia and the other represented by non-motile cilia, also called primary cilia [60][126]. Motile cilia are found on respiratory epithelial cells, ependymal cells of the cerebrospinal fluid spaces, sperm cells, and embryonic node cells during development. Primary cilia act as an antenna for extracellular information and by converting mechanical or chemical stimuli into electrical signals [61][127]. For signal conversion, there are many Ca2+-permeable channels both on the membrane of the cilia and on the basal body, such as polycystin 2 (PC-2), and member 4 of the cation channel subfamily of the transient receptor potential V (TRPV4) (Figure 3 and Figure 4) [62][128].Figure 3. Canonical and non-canonical Wnt pathways. The canonical and noncanonical Wnt pathways are depicted in the figure with the various genes and mechanisms that are activated. In ciliopathies, several studies have shown that this pathway plays an essential role in their onset. Si113 is indicated; an SGK1 kinase inhibitor appears to have an effect on hydrocephalus caused by some ciliopathies.
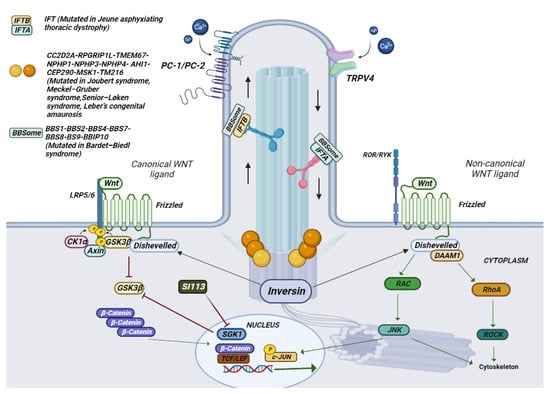
Figure 4. Schematic representation of the composition of a cilium and the various mutated genes in ciliopathies. In the photo it is possible to observe how a cilium is structured in its various components. The different genes mutated in ciliopathies, and their location are represented.