 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ulaganathan Mabalirajan | -- | 3511 | 2023-03-15 11:40:40 | | | |
| 2 | Lindsay Dong | + 7 word(s) | 3518 | 2023-03-16 07:32:44 | | | | |
| 3 | Lindsay Dong | Meta information modification | 3518 | 2023-03-16 07:33:06 | | |
Video Upload Options
The features of allergic asthma are believed to be mediated mostly through the Th2 immune response. In this Th2-dominant concept, the airway epithelium is presented as the helpless victim of Th2 cytokines. Asthma researchers started believing in that the airway epithelium played a crucial role, as alarmins, which are the inducers of type 2 innate lymphoid cell (ILC2), are almost exclusively secreted by the airway epithelium. This underscores the eminence of airway epithelium in asthma pathogenesis. However, the airway epithelium has a bipartite functionality in sustaining healthy lung homeostasis and asthmatic lungs. On the one hand, the airway epithelium maintains lung homeostasis against environmental irritants/pollutants with the aid of its various armamentaria, including its chemosensory apparatus and detoxification system. Alternatively, it induces an ILC2-mediated type 2 immune response through alarmins to amplify the inflammatory response. However, the available evidence indicates that restoring epithelial health may attenuate asthmatic features.
1. Barrier Function of Airway Epithelium
1.1. Importance of Epithelial Barrier in Maintaining Homeostasis
1.2. Anatomical Barrier Role of Airway Epithelium
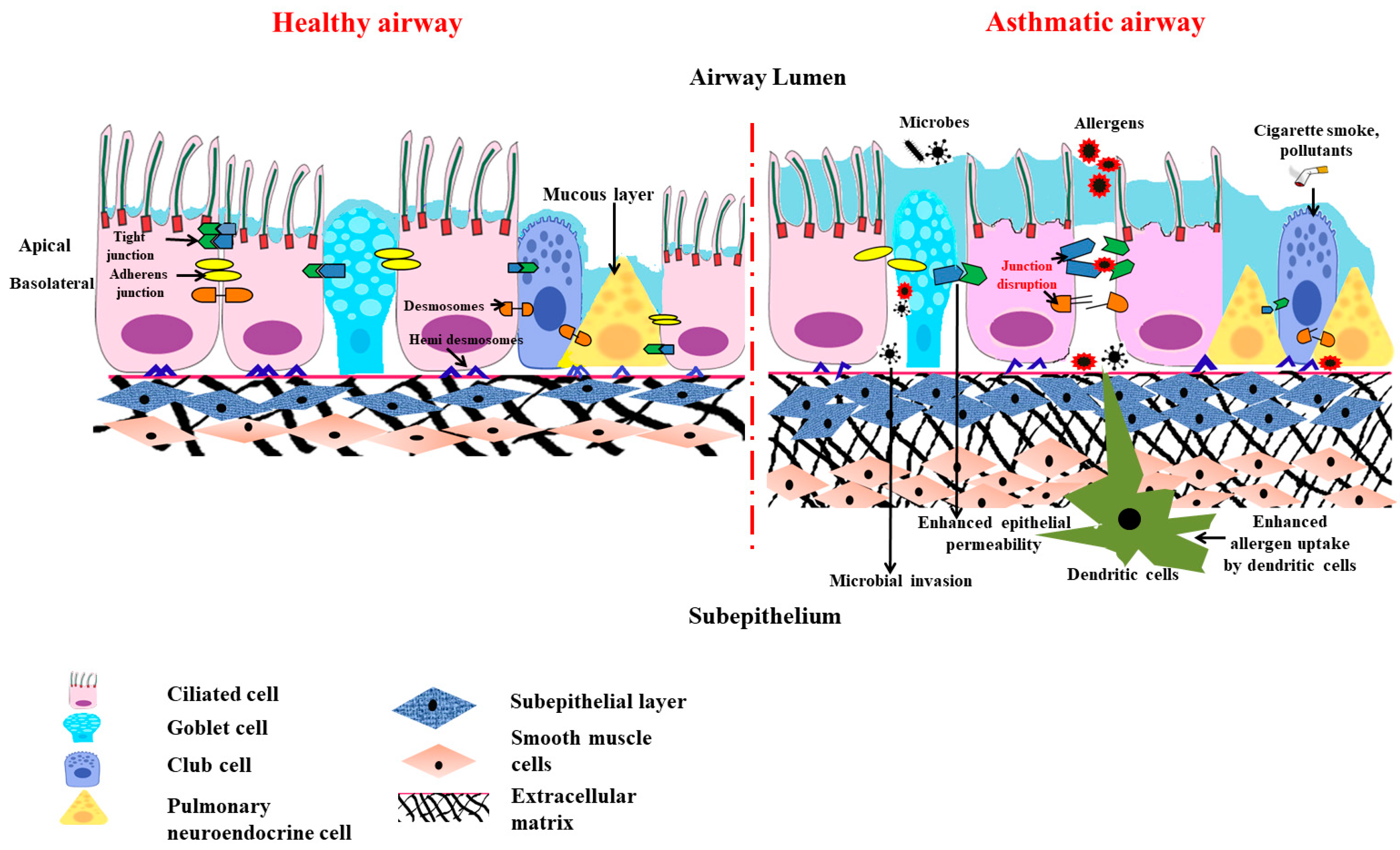
1.3. Chemical Barrier Role of Airway Epithelium
1.4. Physiological Barrier in Airway
1.5. Special Cellular Machinery in Airway Epithelial Barrier
1.6. Barrier Function of Airway Epithelium against Air Pollutants and Pathogens
2. The Victim Role of Airway Epithelium in Asthma Pathogenesis
2.1. Role of Th2 Cytokines in Airway Epithelial Barrier Dysfunction
2.2. Mitochondrial Dysfunction in Asthmatic Airway Epithelium
The earlier clinical study found the presence of an increased number of mitochondria with altered structures in asthmatic children [32]. Later, a lab demonstrated the involvement of mitochondrial dysfunction in asthma pathogenesis [33]. The lab also demonstrated a reduction in the cytochrome c oxidase (COX), which is the primary enzyme of the electron transport chain (ETC) residing in mitochondria’s inner mitochondrial membrane (IMM), which transfers electrons from the cytochrome c to oxygen in the lungs of mice with allergic airway inflammation.
While it is known that both IL-4 and IL-13 promote IgE class switching through STAT-6, they also induce an enzyme: 12/15-lipoxygenase (12/15-LOX, also called 15-lipooxygenase in humans) [34]. Even though the role of 5-lipoxygenase (5-LOX) is well established in asthma pathogenesis, the detailed role of 12/15-lipoxygenase was demonstrated. 12/15-LOX is one of the enzymes responsible for cellular suicide via the programmed disappearance of mitochondria and other cellular organelles from the reticulocytes and for immature fibroblasts converting into red blood cells and mature fibroblasts, respectively [35][36][37]. This is essential for the uninterrupted functions of red blood cells and mature fibroblasts, as the presence of organelles in these cells disturbs their functions, such as effective gas exchange and clear vision, respectively. Though it was known earlier that 12/15-LOX was increased in asthmatic lungs [38][39], its role in mitochondrial dysfunction was not known. The mere overexpression of 12/15-LOX in naïve mice causes mitochondrial dysfunction, along with the development of asthma-like features, indicating the pathogenetic role of 12/15-LOX [40].
3. Governing/Immune Role of Airway Epithelium in Allergic Airway Inflammation
3.1. Less Dominant Role of Inflammation in Causing Epithelial Barrier Dysfunction
3.2. Less Dominant Role of Inflammation in Causing Mitochondrial Dysfunction in Airway Epithelia
3.3. Airway Epithelium Induces ILC2-Mediated Type 2 Immune Response through Alarmins
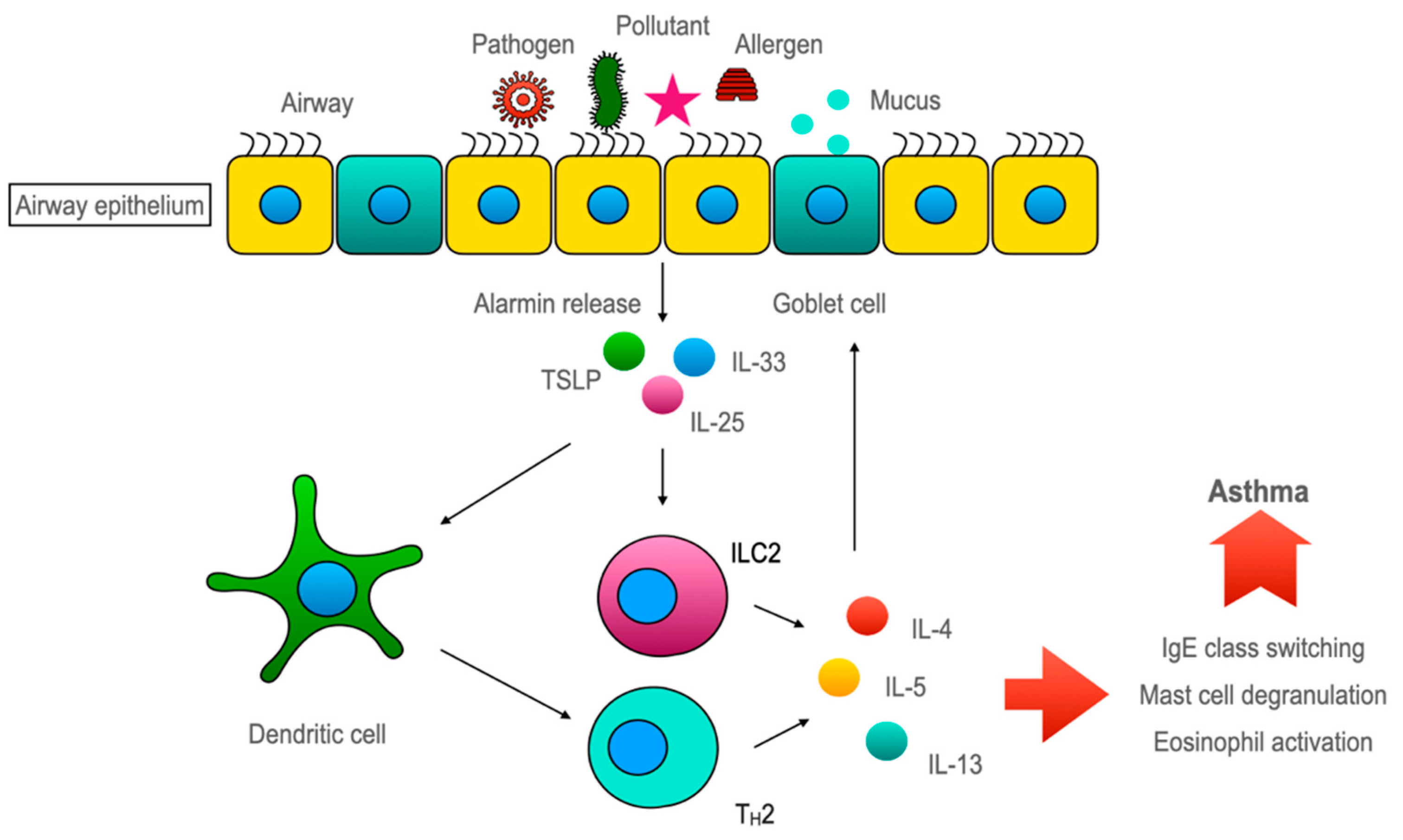
4. Conclusions
References
- Sparr, E.; Millecamps, D.; Isoir, M.; Burnier, V.; Larsson, Å.; Cabane, B. Controlling the hydration of the skin though the application of occluding barrier creams. J. R. Soc. Interface 2012, 10, 20120788.
- Guillot, L.; Nathan, N.; Tabary, O.; Thouvenin, G.; Le Rouzic, P.; Corvol, H.; Amselem, S.; Clement, A. Alveolar epithelial cells: Master regulators of lung homeostasis. Int. J. Biochem. Cell Biol. 2013, 45, 2568–2573.
- Georas, S.N.; Rezaee, F. Epithelial barrier function: At the front line of asthma immunology and allergic airway inflammation. J. Allergy Clin. Immunol. 2014, 134, 509–520.
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 2008, 1778, 660–669.
- Overgaard, C.E.; Mitchell, L.A.; Koval, M. Roles for claudins in alveolar epithelial barrier function. Ann. N. Y. Acad. Sci. 2012, 1257, 167–174.
- Soini, Y. Claudins in lung diseases. Respir. Res. 2011, 12, 70.
- Wittekindt, O.H. Tight junctions in pulmonary epithelia during lung inflammation. Pflug. Arch 2017, 469, 135–147.
- Niessen, C.M.; Gottardi, C.J. Molecular components of the adherens junction. Biochim. Biophys. Acta 2008, 1778, 562–571.
- Fu, R.; Jiang, X.; Li, G.; Zhu, Y.; Zhang, H. Junctional complexes in epithelial cells: Sentinels for extracellular insults and intracellular homeostasis. FEBS J. 2022, 289, 7314–7333.
- Mitamura, Y.; Ogulur, I.; Pat, Y.; Rinaldi, A.O.; Ardicli, O.; Cevhertas, L.; Brüggen, M.C.; Traidl-Hoffmann, C.; Akdis, M.; Akdis, C.A. Dysregulation of the epithelial barrier by environmental and other exogenous factors. Contact Dermat. 2021, 85, 615–626.
- Luissint, A.C.; Parkos, C.A.; Nusrat, A. Inflammation and the Intestinal Barrier: Leukocyte-Epithelial Cell Interactions, Cell Junction Remodeling, and Mucosal Repair. Gastroenterology 2016, 151, 616–632.
- Whitsett, J.A.; Alenghat, T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol. 2015, 16, 27–35.
- Kaushik, M.S.; Chakraborty, S.; Veleri, S.; Kateriya, S. Mucociliary Respiratory Epithelium Integrity in Molecular Defense and Susceptibility to Pulmonary Viral Infections. Biology 2021, 10, 95.
- Zhang, N.; Van Crombruggen, K.; Gevaert, E.; Bachert, C. Barrier function of the nasal mucosa in health and type-2 biased airway diseases. Allergy 2016, 71, 295–307.
- Leiva-Juárez, M.M.; Kolls, J.K.; Evans, S.E. Lung epithelial cells: Therapeutically inducible effectors of antimicrobial defense. Mucosal Immunol. 2018, 11, 21–34.
- Gu, X.; Karp, P.H.; Brody, S.L.; Pierce, R.A.; Welsh, M.J.; Holtzman, M.J.; Ben-Shahar, Y. Chemosensory functions for pulmonary neuroendocrine cells. Am. J. Respir. Cell Mol. Biol. 2014, 50, 637–646.
- Gu, Q.; Lee, L.Y. Neurophysiology: Neural Control of Airway Smooth Muscle. In Encyclopedia of Respiratory Medicine, Four-Volume Set 2006; Laurent, G.J., Shapiro, S.D., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; pp. 138–145.
- Cutz, E.; Yeger, H.; Pan, J. Pulmonary neuroendocrine cell system in pediatric lung disease-recent advances. Pediatr. Dev. Pathol. Off. J. Soc. Pediatr. Pathol. Paediatr. Pathol. Soc. 2007, 10, 419–435.
- Song, H.; Yao, E.; Lin, C.; Gacayan, R.; Chen, M.H.; Chuang, P.T. Functional characterization of pulmonary neuroendocrine cells in lung development, injury, and tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 17531–17536.
- Noguchi, M.; Furukawa, K.T.; Morimoto, M. Pulmonary neuroendocrine cells: Physiology, tissue homeostasis and disease. Dis. Model Mech. 2020, 13, dmm046920.
- Gu, Q.; Lee, l.-y. Neural Control of Airway Smooth Muscle. World J. Anesthesiol. 2020.
- Guarnieri, M.; Balmes, J.R. Outdoor air pollution and asthma. Lancet 2014, 383, 1581–1592.
- Toskala, E.; Kennedy, D.W. Asthma risk factors. Int. Forum. Allergy Rhinol. 2015, 5 (Suppl. 1), S11–S16.
- Tiotiu, A.I.; Novakova, P.; Nedeva, D.; Chong-Neto, H.J.; Novakova, S.; Steiropoulos, P.; Kowal, K. Impact of Air Pollution on Asthma Outcomes. Int. J. Environ. Res. Public Health 2020, 17, 6212.
- León, B.; Ballesteros-Tato, A. Modulating Th2 Cell Immunity for the Treatment of Asthma. Front. Immunol. 2021, 12, 637948.
- Barnes, P.J. Th2 cytokines and asthma: An introduction. Respir. Res. 2001, 2, 64.
- Saatian, B.; Rezaee, F.; Desando, S.; Emo, J.; Chapman, T.; Knowlden, S.; Georas, S.N. Interleukin-4 and interleukin-13 cause barrier dysfunction in human airway epithelial cells. Tissue Barriers 2013, 1, e24333.
- Hellings, P.W.; Steelant, B. Epithelial barriers in allergy and asthma. J. Allergy Clin. Immunol. 2020, 145, 1499–1509.
- Burgess, J.K.; Jonker, M.R.; Berg, M.; Ten Hacken, N.T.; Meyer, K.B.; van den Berge, M.; Nawijn, M.C.; Heijink, I.H. Periostin: Contributor to abnormal airway epithelial function in asthma? Eur. Respir. J. 2021, 57, 2001286.
- Schmidt, H.; Braubach, P.; Schilpp, C.; Lochbaum, R.; Neuland, K.; Thompson, K.; Jonigk, D.; Frick, M.; Dietl, P.; Wittekindt, O.H. IL-13 Impairs Tight Junctions in Airway Epithelia. Int. J. Mol. Sci. 2019, 20, 3222.
- Amin, K.; Janson, C.; Bystrom, J. Role of eosinophil granulocytes in allergic airway inflammation endotypes. Scand. J. Immunol. 2016, 84, 75–85.
- Konrádová, V.; Čopová, C.; Suková, B.; Houštěk, J. Ultrastructure of the bronchial epithelium in three children with asthma. Pediatr. Pulmonol. 1985, 1, 182–187.
- Mabalirajan, U.; Dinda, A.K.; Kumar, S.; Roshan, R.; Gupta, P.; Sharma, S.K.; Ghosh, B. Mitochondrial structural changes and dysfunction are associated with experimental allergic asthma. J. Immunol. 2008, 181, 3540–3548.
- Heydeck, D.; Thomas, L.; Schnurr, K.; Trebus, F.; Thierfelder, W.E.; Ihle, J.N.; Kühn, H. Interleukin-4 and -13 induce upregulation of the murine macrophage 12/15-lipoxygenase activity: Evidence for the involvement of transcription factor STAT6. Blood 1998, 92, 2503–2510.
- Rapoport, S.M.; Schewe, T.; Wiesner, R.; Halangk, W.; Ludwig, P.; JanickeHöhne, M.; Tannert, C.; Hiebsch, C.; Klatt, D. The lipoxygenase of reticulocytes. Purification, characterization and biological dynamics of the lipoxygenase; its identity with the 96 respiratory inhibitors of the reticulocyte. Eur. J. Biochem. 1979, 96, 545–561.
- Watson, A.; Doherty, F.J. Calcium promotes membrane association of reticulocyte 15-lipoxygenase. Biochem. J. 1994, 298, 377–383.
- van Leyen, K.; Duvoisin, R.M.; Engelhardt, H.; Wiedmann, M. A function for lipoxygenase in programmed organelle degradation. Nature 1998, 395, 392–395.
- Chu, H.W.; Balzar, S.; Westcott, J.Y.; Trudeau, J.B.; Sun, Y.; Conrad, D.J.; Wenzel, S.E. Expression and activation of 15-lipoxygenase pathway in severe asthma: Relationship to eosinophilic phenotype and collagen deposition. Clin. Exp. Allergy 2002, 32, 1558–1565.
- Chanez, P.; Bonnans, C.; Chavis, C.; Vachier, I. 15-lipoxygenase: A Janus enzyme? Am. J. Respir. Cell Mol. Biol. 2002, 27, 655–658.
- Mabalirajan, U.; Rehman, R.; Ahmad, T.; Kumar, S.; Leishangthem, G.D.; Singh, S.; Dinda, A.K.; Biswal, S.; Agrawal, A.; Ghosh, B. 12/15-lipoxygenase expressed in non-epithelial cells causes airway epithelial injury in asthma. Sci. Rep. 2013, 3, 1540.
- Heijink, I.H.; Kuchibhotla, V.N.S.; Roffel, M.P.; Maes, T.; Knight, D.A.; Sayers, I.; Nawijn, M.C. Epithelial cell dysfunction, a major driver of asthma development. Allergy 2020, 75, 1902–1917.
- Aghapour, M.; Ubags, N.D.; Bruder, D.; Hiemstra, P.S.; Sidhaye, V.; Rezaee, F.; Heijink, I.H. Role of air pollutants in airway epithelial barrier dysfunction in asthma and COPD. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2022, 31, 210112.
- Loxham, M.; Davies, D.E. Phenotypic and genetic aspects of epithelial barrier function in asthmatic patients. J. Allergy Clin. Immunol. 2017, 139, 1736–1751.
- Runswick, S.; Mitchell, T.; Davies, P.; Robinson, C.; Garrod, D.R. Pollen proteolytic enzymes degrade tight junctions. Respirology 2007, 12, 834–842.
- Guryanova, S.V.; Gigani, O.B.; Gudima, G.O.; Kataeva, A.M.; Kolesnikova, N.V. Dual Effect of Low-Molecular-Weight Bioregulators of Bacterial Origin in Experimental Model of Asthma. Life 2022, 12, 192.
- Aguilera-Aguirre, L.; Bacsi, A.; Saavedra-Molina, A.; Kurosky, A.; Sur, S.; Boldogh, I. Mitochondrial dysfunction increases allergic airway inflammation. J. Immunol. 2009, 183, 5379–5387.
- Barton, J.L.; Berg, T.; Didon, L.; Nord, M. The pattern recognition receptor Nod1 activates CCAAT/enhancer binding protein beta signalling in lung epithelial cells. Eur. Respir. J. 2007, 30, 214–222.
- Farkas, L.; Stoelcker, B.; Jentsch, N.; Heitzer, S.; Pfeifer, M.; Schulz, C. Muramyldipeptide modulates CXCL-8 release of BEAS-2B cells via NOD2. Scand. J. Immunol. 2008, 68, 315–322.
- Kauffman, H.F. Innate immune responses to environmental allergens. Clin. Rev. Allergy Immunol. 2006, 30, 129–140.
- Kato, A.; Favoreto, S.; Avila, P.C.; Schleimer, R.P. TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J. Immunol. 2007, 179, 1080–1087.
- Hammad, H.; Lambrecht, B.N. Dendritic cells and epithelial cells: Linking innate and adaptive immunity in asthma. Nat. Reviews Immunol. 2008, 8, 193–204.
- Oppenheim, J.J.; Yang, D. Alarmins: Chemotactic activators of immune responses. Curr. Opin. Immunol. 2005, 17, 359–365.
- Licona-Limón, P.; Kim, L.K.; Palm, N.W.; Flavell, R.A. TH2, allergy and group 2 innate lymphoid cells. Nat. Immunol. 2013, 14, 536–542.
- Smithgall, M.D.; Comeau, M.R.; Yoon, B.R.; Kaufman, D.; Armitage, R.; Smith, D.E. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int. Immunol. 2008, 20, 1019–1030.
- Gauvreau, G.M.; Bergeron, C.; Boulet, L.P.; Cockcroft, D.W.; Côté, A.; Davis, B.E.; Leigh, R.; Myers, I.; O’Byrne, P.M.; Sehmi, R. Sounding the alarmins-The role of alarmin cytokines in asthma. Allergy 2022, 78, 402–417.

