 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yue Wang | -- | 2701 | 2023-03-13 16:20:12 | | | |
| 2 | Lindsay Dong | Meta information modification | 2701 | 2023-03-14 07:53:06 | | | | |
| 3 | Lindsay Dong | Meta information modification | 2701 | 2023-03-14 07:54:05 | | | | |
| 4 | Lindsay Dong | Meta information modification | 2701 | 2023-03-15 07:04:34 | | |
Video Upload Options
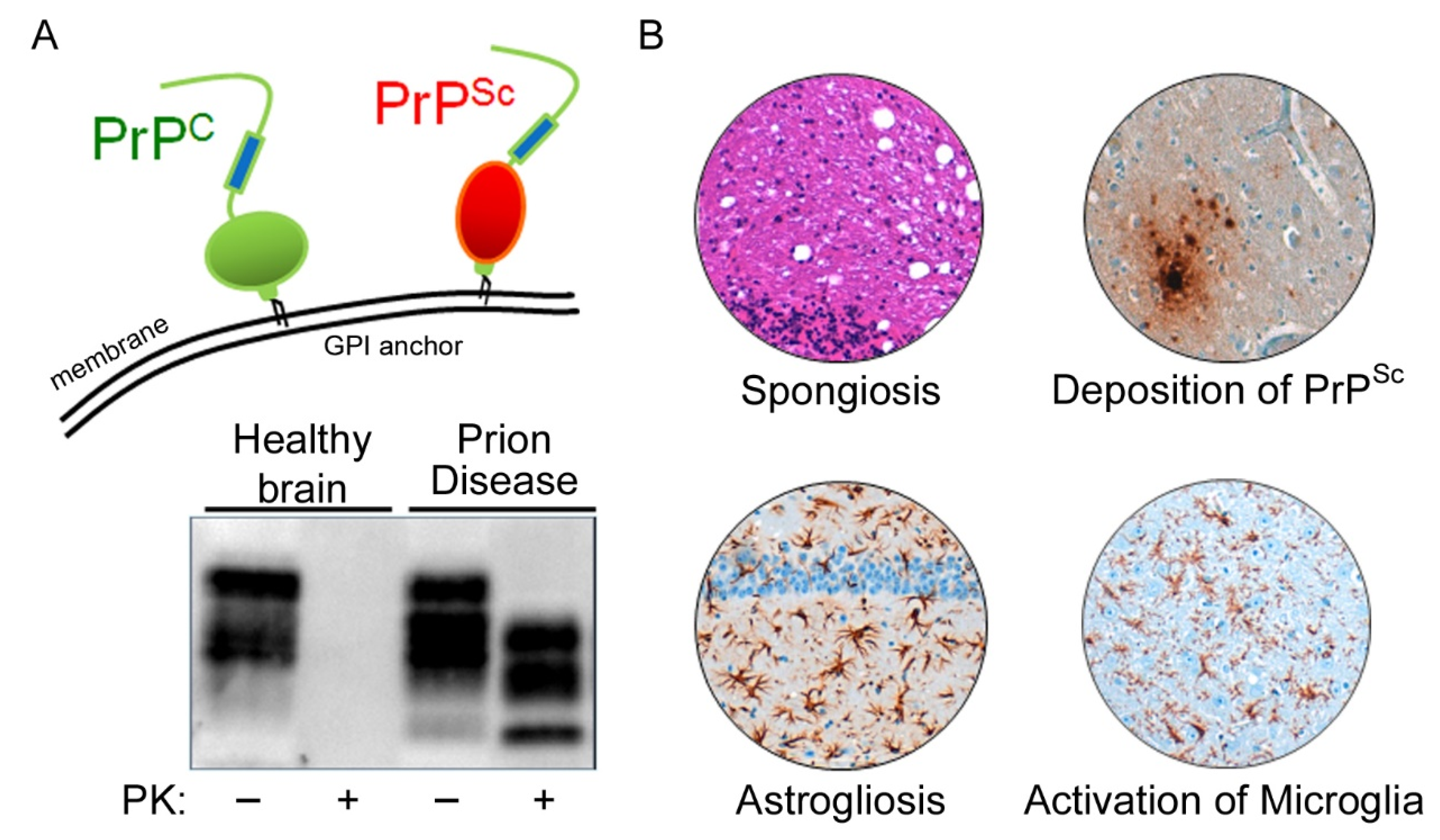
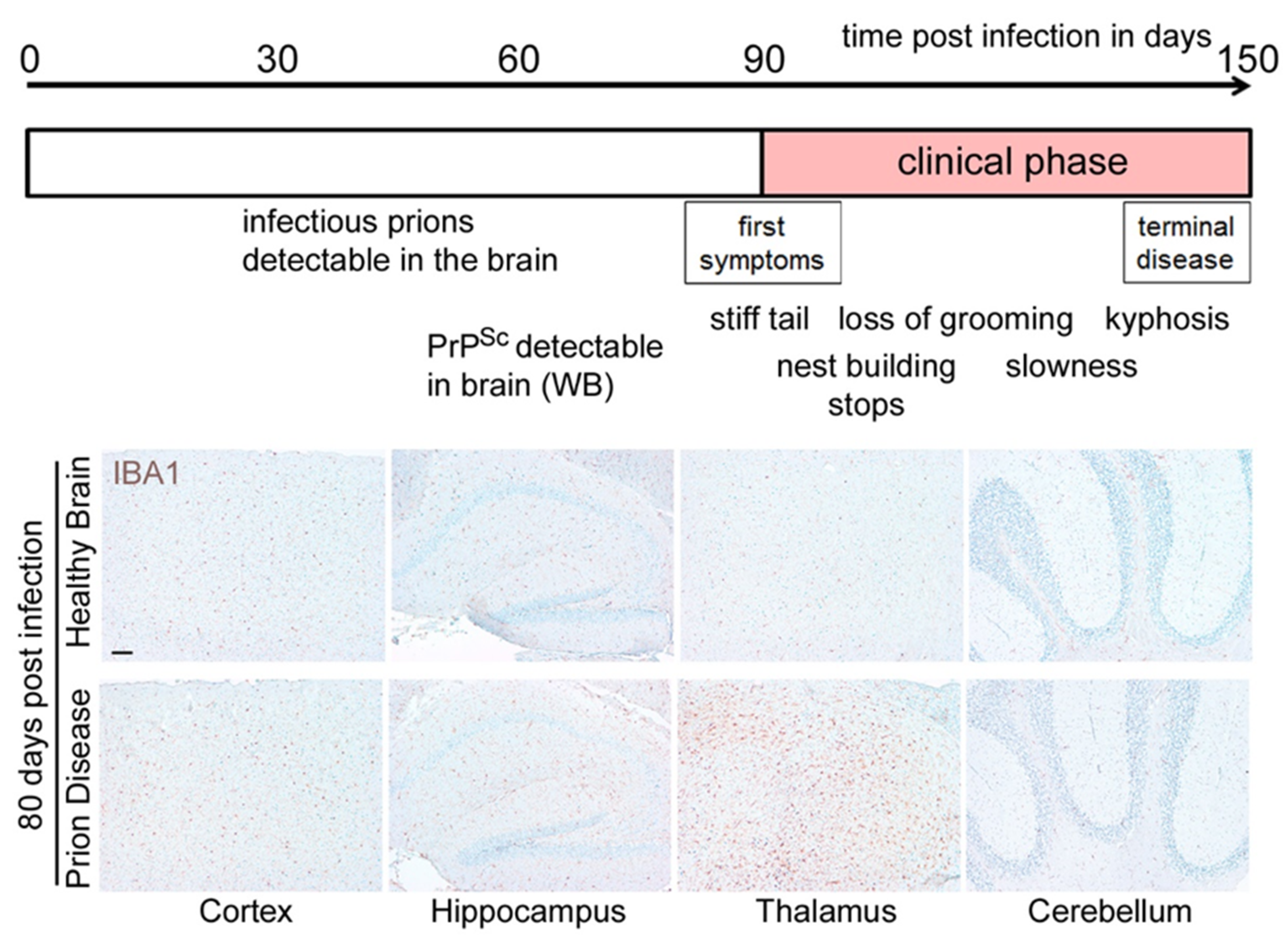
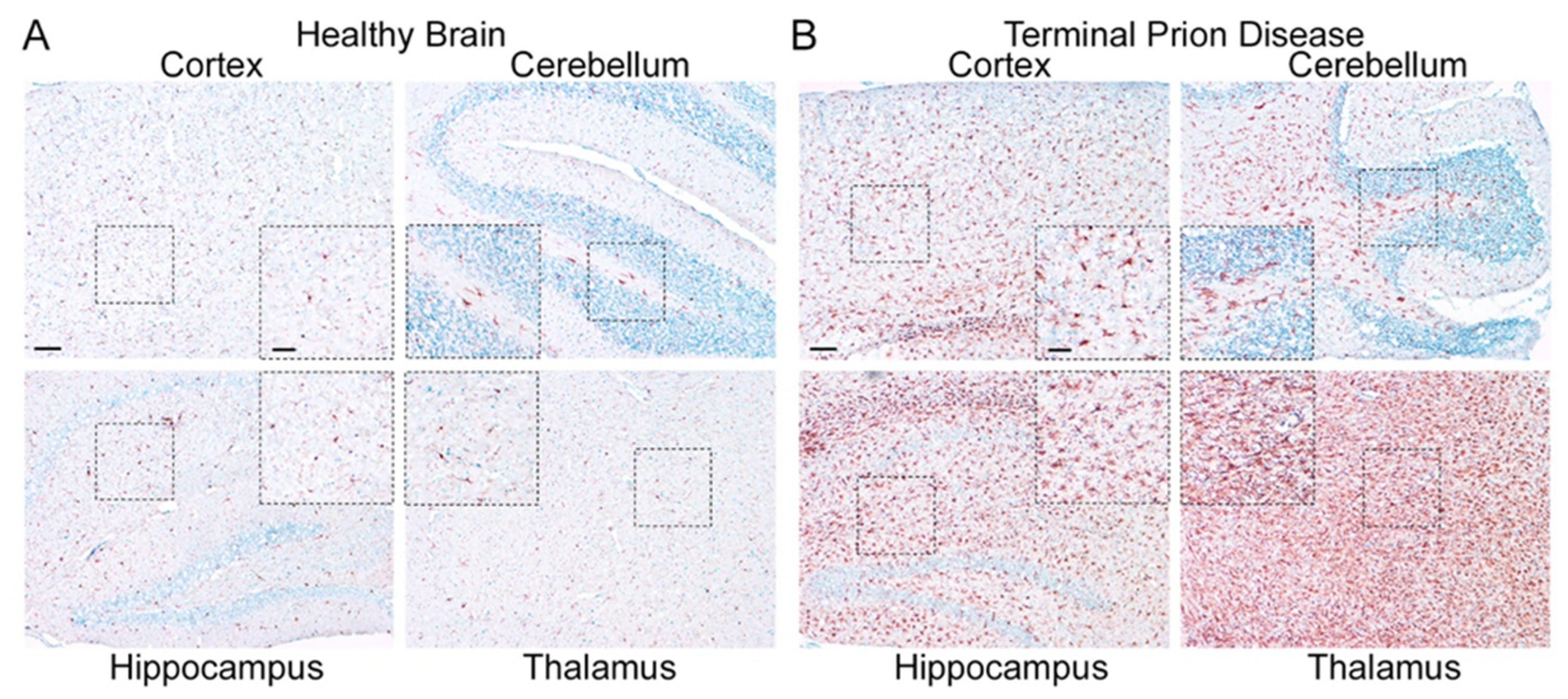
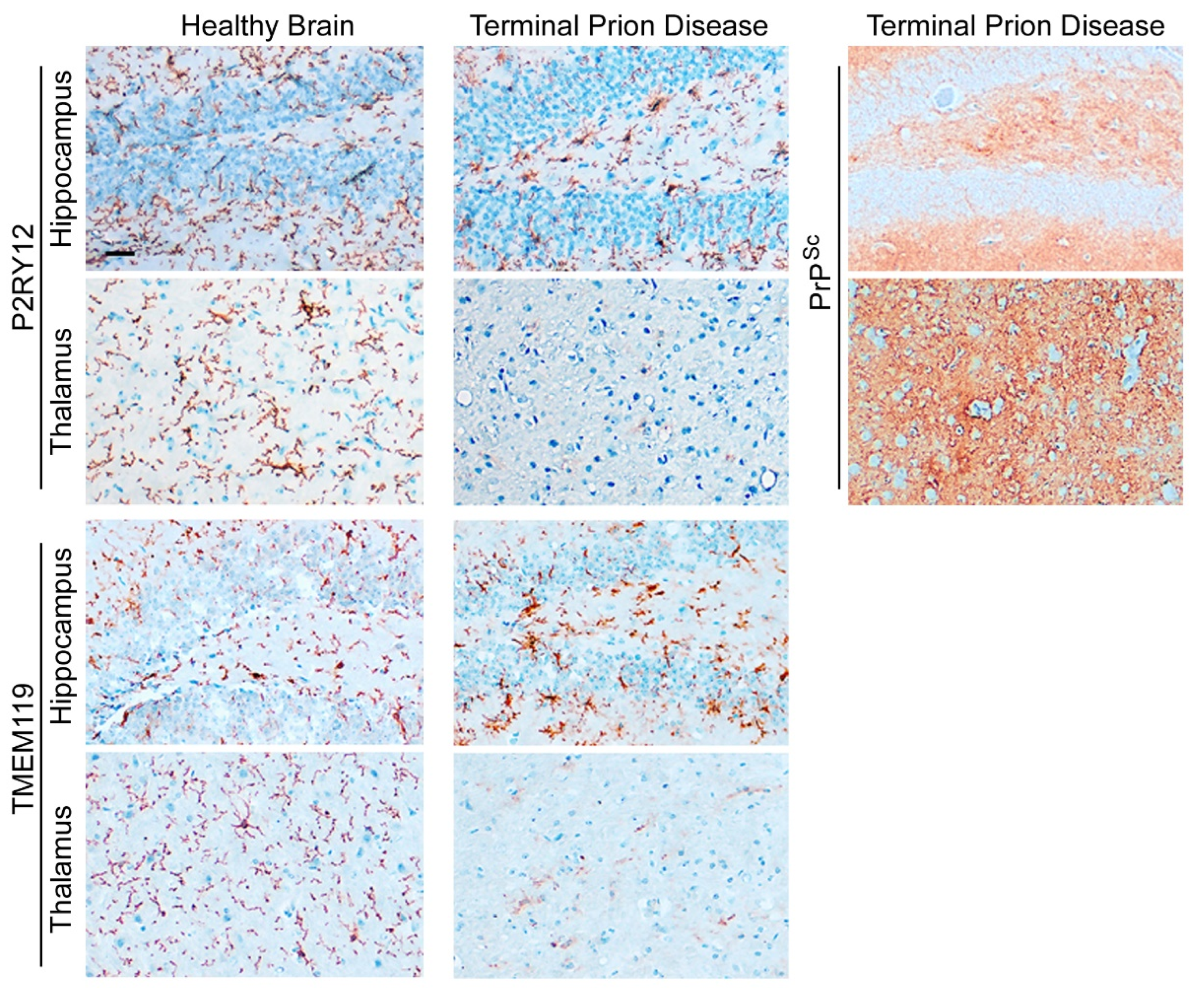
Prion diseases are neurodegenerative diseases that affect humans and animals. They are always fatal and, to date, no treatment exists. The hallmark of prion disease pathophysiology is the misfolding of an endogenous protein, the cellular prion protein (PrPC), into its disease-associated isoform PrPSc. Besides the aggregation and deposition of misfolded PrPSc, prion diseases are characterized by spongiform lesions and the activation of astrocytes and microglia. Microglia are the innate immune cells of the brain. Activated microglia and astrocytes represent a common pathological feature in neurodegenerative disorders. Microglia are dysregulated in neurodegenerative diseases. However, the loss of the microglia homeostatic signature might contribute to disease progression.
1. Introduction
2. Loss of Homeostatic Microglia Signature in Prion Diseases
2.1. Microglia Are Dysregulated and May Have a Dual Role in Prion Diseases
2.2. Homeostatic Microglia: Guardians of the Physiological State
2.3. Human Microglia and Their Regional Heterogeneity
References
- Wells, G.A.; Scott, A.C.; Johnson, C.T.; Gunning, R.F.; Hancock, R.D.; Jeffrey, M.; Dawson, M.; Bradley, R. A novel progressive spongiform encephalopathy in cattle. Vet. Rec. 1987, 121, 419–420.
- Ducrot, C.; Arnold, M.; de Koeijer, A.; Heim, D.; Calavas, D. Review on the epidemiology and dynamics of BSE epidemics. Vet. Res. 2008, 39, 15.
- Sigurdson, C.J.; Mathiason, C.K.; Perrott, M.R.; Eliason, G.A.; Spraker, T.R.; Glatzel, M.; Manco, G.; Bartz, J.C.; Miller, M.W.; Hoover, E.A. Experimental chronic wasting disease (CWD) in the ferret. J. Comp. Pathol. 2008, 138, 189–196.
- Creutzfeldt, H.G. Über eine eigenartige herdförmige Erkrankung des Zentralnervensystems. Z. Ges. Neurol. Psychiatr. 1920, 57, 1–19.
- Jakob, A. Über eigenartige Erkrankungen des Zentralnervensystems mit bemerkenswertem anatomischem Befunde. (Spastische Pseudosklerose-Encephalomyelopathie mit disseminierten Degenerationsherden). Z. ges. Neurol. Psychiatr. 1921, 64, 147–228.
- Gajdusek, D.C.; Reid, L.H. Studies on kuru. IV. The kuru pattern in Moke, a representative Fore village. Am. J. Trop. Med. Hyg. 1961, 10, 628–638.
- Ironside, J.W. Prion diseases in man. J. Pathol. 1998, 186, 227–234.
- Collins, S.; McLean, C.A.; Masters, C.L. Gerstmann-Straussler-Scheinker syndrome, fatal familial insomnia, and kuru: A review of these less common human transmissible spongiform encephalopathies. J. Clin. Neurosci. 2001, 8, 387–397.
- Kovacs, G.G.; Puopolo, M.; Ladogana, A.; Pocchiari, M.; Budka, H.; van Duijn, C.; Collins, S.J.; Boyd, A.; Giulivi, A.; Coulthart, M.; et al. Genetic prion disease: The EUROCJD experience. Hum. Genet. 2005, 118, 166–174.
- Medori, R.; Tritschler, H.J.; LeBlanc, A.; Villare, F.; Manetto, V.; Chen, H.Y.; Xue, R.; Leal, S.; Montagna, P.; Cortelli, P.; et al. Fatal familial insomnia, a prion disease with a mutation at codon 178 of the prion protein gene. N. Engl. J. Med. 1992, 326, 444–449.
- Glatzel, M.; Rogivue, C.; Ghani, A.; Streffer, J.R.; Amsler, L.; Aguzzi, A. Incidence of Creutzfeldt-Jakob disease in Switzerland. Lancet 2002, 360, 139–141.
- Parchi, P.; Castellani, R.; Capellari, S.; Ghetti, B.; Young, K.; Chen, S.G.; Farlow, M.; Dickson, D.W.; Sima, A.A.F.; Trojanowski, J.Q.; et al. Molecular Basis of Phenotypic Variability in Sporadic Creutzfeldt-Jakob Disease. Ann. Neurol. 1996, 39, 767–778.
- Alperovitch, A.; Zerr, I.; Pocchiari, M.; Mitrova, E.; de Pedro Cuesta, J.; Hegyi, I.; Collins, S.; Kretzschmar, H.; van Duijn, C.; Will, R.G. Codon 129 prion protein genotype and sporadic Creutzfeldt-Jakob disease . Lancet 1999, 353, 1673–1674.
- Hill, A.F.; Desbruslais, M.; Joiner, S.; Sidle, K.C.; Gowland, I.; Collinge, J.; Doey, L.J.; Lantos, P. The same prion strain causes vCJD and BSE . Nature 1997, 389, 448–450.
- Will, R.G.; Ironside, J.W.; Zeidler, M.; Cousens, S.N.; Estibeiro, K.; Alperovitch, A.; Poser, S.; Pocchiari, M.; Hofman, A.; Smith, P.G. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 1996, 347, 921–925.
- Sanchez-Juan, P.; Bishop, M.T.; Kovacs, G.G.; Calero, M.; Aulchenko, Y.S.; Ladogana, A.; Boyd, A.; Lewis, V.; Ponto, C.; Calero, O.; et al. A genome wide association study links glutamate receptor pathway to sporadic Creutzfeldt-Jakob disease risk. PLoS ONE 2014, 10, e0123654.
- Jones, E.; Hummerich, H.; Vire, E.; Uphill, J.; Dimitriadis, A.; Speedy, H.; Campbell, T.; Norsworthy, P.; Quinn, L.; Whitfield, J.; et al. Identification of novel risk loci and causal insights for sporadic Creutzfeldt-Jakob disease: A genome-wide association study. Lancet Neurol. 2020, 19, 840–848.
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144.
- Stahl, N.; Borchelt, D.R.; Hsiao, K.; Prusiner, S.B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987, 51, 229–240.
- Horiuchi, M.; Caughey, B. Specific binding of normal prion protein to the scrapie form via a localized domain initiates its conversion to the protease-resistant state. EMBO J. 1999, 18, 3193–3203.
- Caughey, B.; Raymond, G.J. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease and phospholipase-sensitive. J. Biol. Chem. 1991, 266, 18217–18223.
- Bueler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347.
- Kimberlin, R.H.; Walker, C.A.; Fraser, H. The genomic identity of different strains of mouse scrapie is expressed in hamsters and preserved on reisolation in mice. J. Gen. Virol. 1989, 70, 2017–2025.
- Cunningham, C.; Deacon, R.M.; Chan, K.; Boche, D.; Rawlins, J.N.; Perry, V.H. Neuropathologically distinct prion strains give rise to similar temporal profiles of behavioral deficits. Neurobiol. Dis 2005, 18, 258–269.
- Aguzzi, A.; Nuvolone, M.; Zhu, C. The immunobiology of prion diseases. Nat. Rev. Immunol. 2013, 13, 888–902.
- Betmouni, S.; Perry, V.H.; Gordon, J.L. Evidence for an early inflammatory response in the central nervous system of mice with scrapie. Neuroscience 1996, 74, 1–5.
- Giese, A.; Brown, D.R.; Groschup, M.H.; Feldmann, C.; Haist, I.; Kretzschmar, H.A. Role of microglia in neuronal cell death in prion disease. Brain Pathol. 1998, 8, 449–457.
- Perry, V.H.; Cunningham, C.; Boche, D. Atypical inflammation in the central nervous system in prion disease. Curr. Opin. Neurol. 2002, 15, 349–354.
- Sorce, S.; Nuvolone, M.; Russo, G.; Chincisan, A.; Heinzer, D.; Avar, M.; Pfammatter, M.; Schwarz, P.; Delic, M.; Muller, M.; et al. Genome-wide transcriptomics identifies an early preclinical signature of prion infection. PLoS Pathog. 2020, 16, e1008653.
- Prinz, M.; Heikenwalder, M.; Schwarz, P.; Takeda, K.; Akira, S.; Aguzzi, A. Prion pathogenesis in the absence of Toll-like receptor signalling. EMBO Rep. 2003, 4, 195–199.
- Slota, J.A.; Medina, S.J.; Frost, K.L.; Booth, S.A. Neurons and Astrocytes Elicit Brain Region Specific Transcriptional Responses to Prion Disease in the Murine CA1 and Thalamus. Front. Neurosci. 2022, 16, 918811.
- Scheckel, C.; Imeri, M.; Schwarz, P.; Aguzzi, A. Ribosomal profiling during prion disease uncovers progressive translational derangement in glia but not in neurons. Elife 2020, 9, e62911.
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845.
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Holscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280.
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543.
- Bruttger, J.; Karram, K.; Wortge, S.; Regen, T.; Marini, F.; Hoppmann, N.; Klein, M.; Blank, T.; Yona, S.; Wolf, Y.; et al. Genetic Cell Ablation Reveals Clusters of Local Self-Renewing Microglia in the Mammalian Central Nervous System. Immunity 2015, 43, 92–106.
- Askew, K.; Li, K.; Olmos-Alonso, A.; Garcia-Moreno, F.; Liang, Y.; Richardson, P.; Tipton, T.; Chapman, M.A.; Riecken, K.; Beccari, S.; et al. Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Rep. 2017, 18, 391–405.
- Tay, T.L.; Mai, D.; Dautzenberg, J.; Fernandez-Klett, F.; Lin, G.; Sagar; Datta, M.; Drougard, A.; Stempfl, T.; Ardura-Fabregat, A.; et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci. 2017, 20, 793–803.
- Gomez-Nicola, D.; Fransen, N.L.; Suzzi, S.; Perry, V.H. Regulation of microglial proliferation during chronic neurodegeneration. J. Neurosci. 2013, 33, 2481–2493.
- Carroll, J.A.; Race, B.; Williams, K.; Striebel, J.; Chesebro, B. Microglia Are Critical in Host Defense against Prion Disease. J. Virol. 2018, 92, 00549.
- Muth, C.; Schrock, K.; Madore, C.; Hartmann, K.; Fanek, Z.; Butovsky, O.; Glatzel, M.; Krasemann, S. Activation of microglia by retroviral infection correlates with transient clearance of prions from the brain but does not change incubation time. Brain Pathol. 2017, 27, 590–602.
- Chouhan, J.K.; Puntener, U.; Booth, S.G.; Teeling, J.L. Systemic Inflammation Accelerates Changes in Microglial and Synaptic Markers in an Experimental Model of Chronic Neurodegeneration. Front. Neurosci. 2021, 15, 760721.
- Pal, R.; Bradford, B.M.; Mabbott, N.A. Innate Immune Tolerance in Microglia Does Not Impact on Central Nervous System Prion Disease. Front. Cell Neurosci. 2022, 16, 918883.
- Zhu, C.; Herrmann, U.S.; Falsig, J.; Abakumova, I.; Nuvolone, M.; Schwarz, P.; Frauenknecht, K.; Rushing, E.J.; Aguzzi, A. A neuroprotective role for microglia in prion diseases. J. Exp. Med. 2016, 213, 1047–1059.
- Aguzzi, A.; Zhu, C. Microglia in prion diseases. J. Clin. Invest. 2017, 127, 3230–3239.
- Obst, J.; Simon, E.; Martin-Estebane, M.; Pipi, E.; Barkwill, L.M.; Gonzalez-Rivera, I.; Buchanan, F.; Prescott, A.R.; Faust, D.; Fox, S.; et al. Inhibition of IL-34 Unveils Tissue-Selectivity and Is Sufficient to Reduce Microglial Proliferation in a Model of Chronic Neurodegeneration. Front. Immunol. 2020, 11, 579000.
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318.
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178.
- Tremblay, M.E.; Lowery, R.L.; Majewska, A.K. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010, 8, e1000527.
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458.
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553.
- Aguzzi, A.; Barres, B.A.; Bennett, M.L. Microglia: Scapegoat, saboteur, or something else? Science 2013, 339, 156–161.
- Prokop, S.; Miller, K.R.; Heppner, F.L. Microglia actions in Alzheimer’s disease. Acta Neuropathol. 2013, 126, 461–477.
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405.
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu Rev. Immunol. 2009, 27, 119–145.
- Riemer, C.; Neidhold, S.; Burwinkel, M.; Schwarz, A.; Schultz, J.; Kratzschmar, J.; Monning, U.; Baier, M. Gene expression profiling of scrapie-infected brain tissue. Biochem. Biophys. Res. Commun. 2004, 323, 556–564.
- Carroll, J.A.; Striebel, J.F.; Race, B.; Phillips, K.; Chesebro, B. Prion infection of mouse brain reveals multiple new upregulated genes involved in neuroinflammation or signal transduction. J. Virol. 2015, 89, 2388–2404.
- Vincenti, J.E.; Murphy, L.; Grabert, K.; McColl, B.W.; Cancellotti, E.; Freeman, T.C.; Manson, J.C. Defining the Microglia Response during the Time Course of Chronic Neurodegeneration. J. Virol. 2015, 90, 3003–3017.
- Hwang, D.; Lee, I.Y.; Yoo, H.; Gehlenborg, N.; Cho, J.H.; Petritis, B.; Baxter, D.; Pitstick, R.; Young, R.; Spicer, D.; et al. A systems approach to prion disease. Mol. Syst. Biol. 2009, 5, 252.
- Hartmann, K.; Sepulveda-Falla, D.; Rose, I.V.L.; Madore, C.; Muth, C.; Matschke, J.; Butovsky, O.; Liddelow, S.; Glatzel, M.; Krasemann, S. Complement 3(+)-astrocytes are highly abundant in prion diseases, but their abolishment led to an accelerated disease course and early dysregulation of microglia. Acta Neuropathol. Commun. 2019, 7, 83.
- Riemer, C.; Schultz, J.; Burwinkel, M.; Schwarz, A.; Mok, S.W.; Gultner, S.; Bamme, T.; Norley, S.; van Landeghem, F.; Lu, B.; et al. Accelerated prion replication in, but prolonged survival times of, prion-infected CXCR3-/- mice. J. Virol. 2008, 82, 12464–12471.
- Sakai, K.; Hasebe, R.; Takahashi, Y.; Song, C.H.; Suzuki, A.; Yamasaki, T.; Horiuchi, M. Absence of CD14 delays progression of prion diseases accompanied by increased microglial activation. J. Virol. 2013, 87, 13433–13445.
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678.
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673.
- Nuvolone, M.; Sorce, S.; Schwarz, P.; Aguzzi, A. Prion pathogenesis in the absence of NLRP3/ASC inflammasomes. PLoS ONE 2015, 10, e0117208.
- Hughes, M.M.; Field, R.H.; Perry, V.H.; Murray, C.L.; Cunningham, C. Microglia in the degenerating brain are capable of phagocytosis of beads and of apoptotic cells, but do not efficiently remove PrPSc, even upon LPS stimulation. Glia 2010, 58, 2017–2030.
- Carroll, J.A.; Race, B.; Williams, K.; Chesebro, B. Toll-like receptor 2 confers partial neuroprotection during prion disease. PLoS ONE 2018, 13, e0208559.
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143.
- Butovsky, O.; Jedrychowski, M.P.; Cialic, R.; Krasemann, S.; Murugaiyan, G.; Fanek, Z.; Greco, D.J.; Wu, P.M.; Doykan, C.E.; Kiner, O.; et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann. Neurol. 2015, 77, 75–99.
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain 2017, 140, 1900–1913.
- Sobue, A.; Komine, O.; Hara, Y.; Endo, F.; Mizoguchi, H.; Watanabe, S.; Murayama, S.; Saito, T.; Saido, T.C.; Sahara, N.; et al. Microglial gene signature reveals loss of homeostatic microglia associated with neurodegeneration of Alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 1.
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635.
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128.
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905.
- Kenkhuis, B.; Somarakis, A.; Kleindouwel, L.R.T.; van Roon-Mom, W.M.C.; Hollt, T.; van der Weerd, L. Co-expression patterns of microglia markers Iba1, TMEM119 and P2RY12 in Alzheimer’s disease. Neurobiol. Dis. 2022, 167, 105684.
- Satoh, J.; Kino, Y.; Asahina, N.; Takitani, M.; Miyoshi, J.; Ishida, T.; Saito, Y. TMEM119 marks a subset of microglia in the human brain. Neuropathology 2016, 36, 39–49.
- Krbot, K.; Hermann, P.; Krbot Skorić, M.; Zerr, I.; Sepulveda-Falla, D.; Goebel, S.; Matschke, J.; Krasemann, S.; Glatzel, M. Distinct microglia profile in Creutzfeldt-Jakob disease and Alzheimer’s disease is independent of disease kinetics. Neuropathology 2018, 38, 591–600.
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Melief, J.; Kooijman, L.; Huitinga, I.; Klooster, J.; Bossers, K.; Hol, E.M. Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol. Aging 2014, 35, 1–14.
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504–516.
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.
- Yang, A.C.; Kern, F.; Losada, P.M.; Agam, M.R.; Maat, C.A.; Schmartz, G.P.; Fehlmann, T.; Stein, J.A.; Schaum, N.; Lee, D.P.; et al. Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature 2021, 595, 565–571.
- Areskeviciute, A.; Litman, T.; Broholm, H.; Melchior, L.C.; Nielsen, P.R.; Green, A.; Eriksen, J.O.; Smith, C.; Lund, E.L. Regional Differences in Neuroinflammation-Associated Gene Expression in the Brain of Sporadic Creutzfeldt-Jakob Disease Patients. Int. J. Mol. Sci. 2020, 22, 140.
- Llorens, F.; Thune, K.; Tahir, W.; Kanata, E.; Diaz-Lucena, D.; Xanthopoulos, K.; Kovatsi, E.; Pleschka, C.; Garcia-Esparcia, P.; Schmitz, M.; et al. YKL-40 in the brain and cerebrospinal fluid of neurodegenerative dementias. Mol. Neurodegener. 2017, 12, 83.
- Parchi, P.; Giese, A.; Capellari, S.; Brown, P.; Schulz-Schaeffer, W.; Windl, O.; Zerr, I.; Budka, H.; Kopp, N.; Piccardo, P.; et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann. Neurol. 1999, 46, 224–233.
- Ironside, J.W.; Ritchie, D.L.; Head, M.W. Phenotypic variability in human prion diseases. Neuropathol. Appl. Neurobiol. 2005, 31, 565–579.
- Budka, H. Neuropathology of prion diseases. Br. Med. Bull. 2003, 66, 121–130.
- Geissen, M.; Krasemann, S.; Matschke, J.; Glatzel, M. Understanding the natural variability of prion diseases. Vaccine 2007, 25, 5631–5636.

