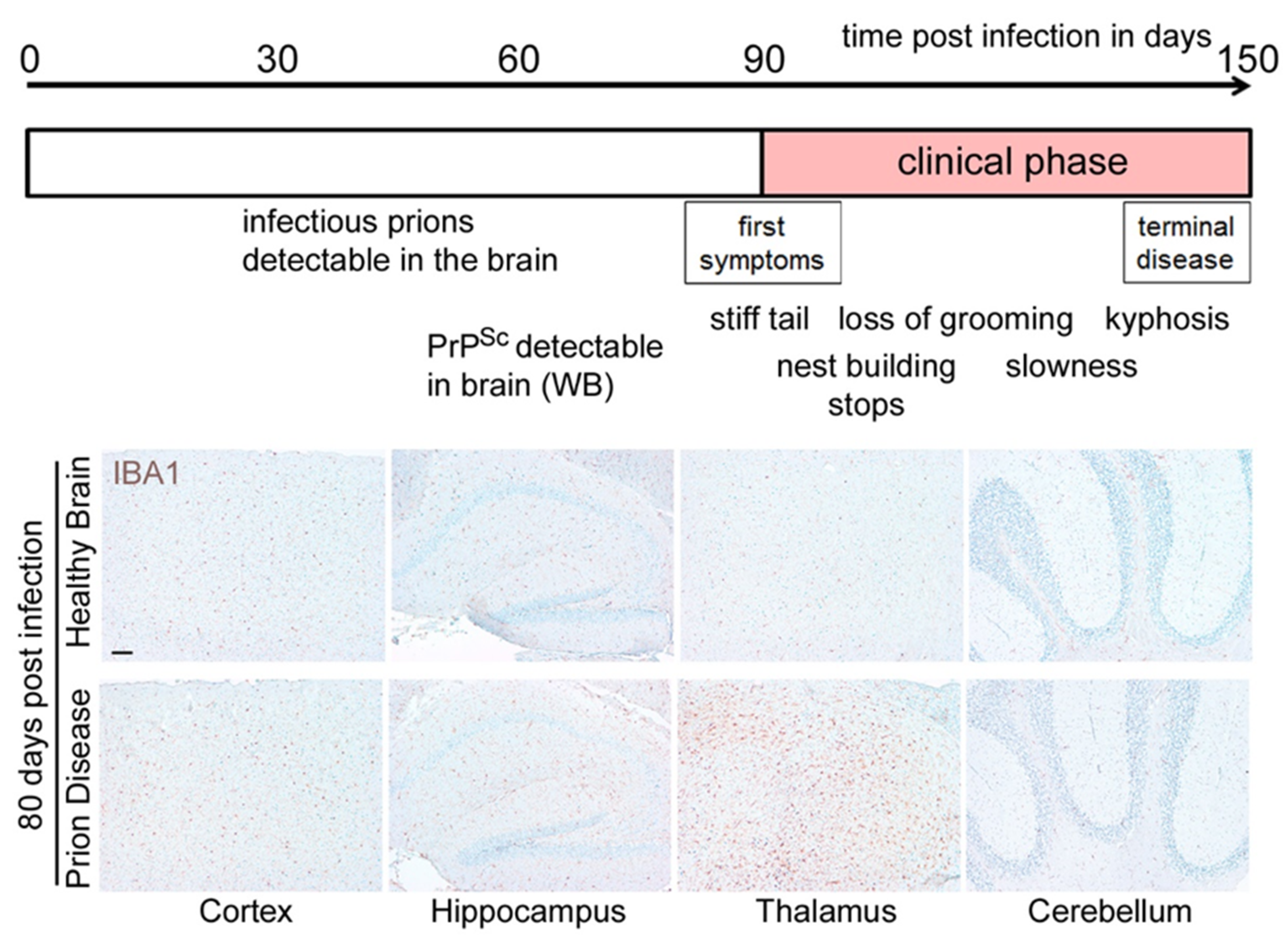
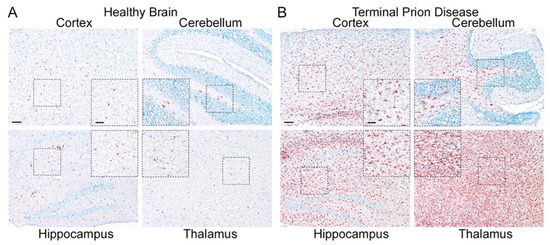
Figure 3. Microglia are highly activated in terminal prion disease. The pan-microglia/macrophage marker IBA1 is shown in mouse brain sections of (
A) mock-infected healthy mice and (
B) RML 5.0 prion-infected mice at a terminal disease state. (
A) Microglia in the healthy brain show a ramified phenotype with a small soma and thin processes in all four regions displayed here (see magnified close-up). (
B) During the terminal prion disease stage, microglia massively proliferate (see overview) and change their morphology towards bigger cell bodies and thicker arms (see close-ups) with a bushy appearance in the hippocampus and an amoeboid phenotype in the thalamus
[39][40][41][45,46,47]. Scale bar: 100 µm; close-up: 50 µm.
Microglia in the healthy brain display a homeostatic morphology characterized by a small cell body and long branching processes which are constantly surveying their environment
[47][61]. Moreover, microglia are involved in several mechanisms regulating brain development and plasticity. They actively shape neuronal connectivity by the removal of excess/neglected synapses, which are formed during development
[48][49][50][62,63,64]. For this, microglia have been shown to directly contact tagged pre- and postsynaptic structures and remove them by phagocytosis. The targeting of unwanted elements for removal involves complement receptors and adaptor proteins, among other signals
[48][50][62,64].
Microglia can react to alterations in the brain environment or the presence of threats to its cellular integrity e.g., invading pathogens and aggregated proteins, but also apoptotic cells or cell debris, by mounting an inflammatory response to restore homeostasis. The latter enables microglia to increase their phagocytic capacity to remove unwanted structures and to release proinflammatory mediators including cytokines and chemokines
[51][65]. Activated microglia represent a common feature of neurodegenerative diseases
[52][53][54][54,66,67]. Activation can be identified by a combination of morphological and immuno-phenotypic changes
[55][68]. Several proinflammatory cytokines are upregulated in the brain during the prion disease course including TNF α, IL 1α, and C1qa
[40][56][57][58][59][60][46,69,70,71,72,73].
Since pro-inflammatory mediators are upregulated slightly before the onset of symptoms, several studies were conducted to investigate the impact of manipulating microglia receptor abundance or cytokine release, and yielded a somewhat mixed outcome. In line with the proposed capacity of microglia to phagocytose PrP
Sc, the knockout of C-X-C chemokine receptor type 3 (CXCR3) led to accelerated PrP
Sc accumulation and increased prion infectivity titers, but prolonged survival in the mouse model
[61][76]. Moreover, these mice developed excessive astrocytosis. The knockout of the cluster of differentiation CD14 also led to a prolonged survival with enhanced microglia activation in two different mouse models
[62][77]. The NLRP3 inflammasome is a multi-molecular complex which can sense heterogeneous pathogen-associated molecular patterns (PAMPs) culminating in the activation of caspase 1 and the release of interleukin IL 1β. It has been shown to play a fundamental role in Alzheimer’s disease (AD) pathophysiology
[63][64][78,79]. In contrast to AD, the knockout of NLRP3 or the adaptor protein ASC does not influence the prion disease course
[65][80]. Interestingly, retroviral infection preceding prion infection in mice led to a stimulation of microglia at early disease time points with a reduction in infectious prions
[41][47], while another stimulator of inflammation, the bacterial lipopolysaccharide (LPS), could not further stimulate their PrP
Sc-degrading function during disease progression
[66][81]. In addition to the upregulation of PAMPs in prion diseases, the transcription of several genes encoding damage-associated molecular pattern (DAMP) proteins and receptors such as Toll-like receptors or proteins of the complement cascade are also increased in the brains of prion-infected mice
[57][67][70,82]. Interestingly, depletion of any of the DAMP receptor genes
Tlr2,
C3ar1, and
C5ar1 in a prion mouse model only led to a slightly increased survival in the
Tlr2-model, while
C3ar1, and
C5ar1 did not influence prion disease course
[67][82].
2.2. Homeostatic Microglia: Guardians of the Physiological State
For decades, microglia research was focused on the inflammatory context and profile. Only recent research has brought into the focus the homeostatic functions of microglia, which—if lost—might be a driver of neurodegeneration in disease
[68][69][70][71][72][55,88,89,90,91]. However, the identification of microglia-specific profiles was complicated by the lack of understanding of whether and how brain-resident microglia functionally differ from peripheral myeloid cells which might enter the brain in certain instances
[73][92]. Only recently were the molecular and functional characteristics of murine bona fide homeostatic microglia identified by different groups using a set of novel methodologies such as quantitative proteomics or RNA sequencing
[69][74][75][88,93,94]. Thus, a unique transcriptional expression signature of homeostatic microglia was identified which allowed for their differentiation from peripheral myeloid cells. Homeostatic microglia are characterized by the expression of specific markers such as
Tmem119,
Hexb,
Gpr34,
P2ry12,
Olfml3, and
Tgfbr1. Several of these genes are also expressed on human microglia, including the P2Y purinoceptor 12 (
P2RY12)
[76][95] and the transmembrane protein
TMEM119 [77][96]. The development of robust tools including microglia-specific antibodies followed soon after.
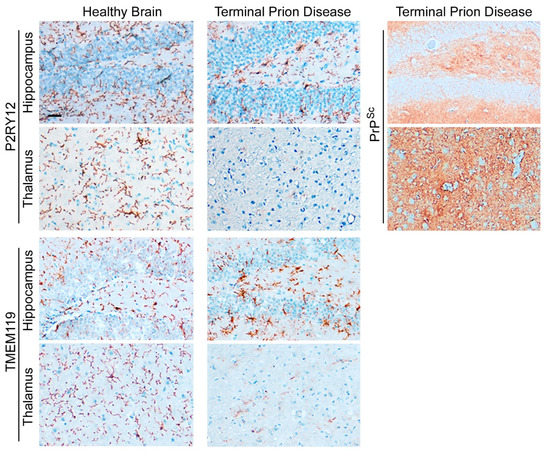
The abundance of homeostatic microglia proteins such as TMEM119 and P2RY12 is significantly reduced in terminal prion disease (
Figure 4)
[41][60][78][47,73,97]. In the healthy brain, microglia show a ramified morphology with a small cell body and thin processes that are especially visible after staining with P2RY12
[68][69][70][71][72][55,88,89,90,91]. In contrast, both markers are affected in terminal prion disease. While the homeostatic markers are still abundant in the hippocampus, this signature is almost completely lost in the thalamus, an effect which is especially severe for P2RY12 (
Figure 4)
[41][60][78][47,73,97].
Figure 4. Loss of microglial homeostatic phenotype in terminal prion disease. The microglia-specific homeostasis markers P2RY12 and TMEM119 show a ramified morphology of microglia in the healthy brain
[41][60][78][47,73,97]. In terminal prion disease, this homeostatic microglia signature is lost in a region-dependent manner
[41][60][78][47,73,97]. While the activated microglia in the dentate gyrus of the hippocampus display a bushy morphology with thick and retracted processes and still express both markers, microglia in the thalamus (posterior complex) have almost completely lost the expression of the markers, especially P2RY12. In the RML-prion mouse model, the deposition of PrP
Sc-specific staining is stronger in the thalamus than in the hippocampus in terminal prion disease. Scale bar: 25 µm.
2.3. Human Microglia and Their Regional Heterogeneity
For decades it has been noted, mainly based on morphological data from immunohistochemical staining, that the microglia population within the brain is not uniform. Over the last ten years, microglia heterogeneity has been studied in murine models, determining that microglia have distinct region-dependent transcriptional identities and that they age in a regionally variable manner
[79][80][109,110]. Recent advances in single-cell sequencing have shown the diversity and regional heterogeneity of murine brain microglia in more detail
[81][111].
Although a variety of new molecular tools, such as nuclear sequencing, are now available to study expression profiles on a single-cell basis in the human brain in health and disease
[82][113], the transmissible nature of the fatal yet untreatable prion disorders and their respective biosafety issues limit their use in the study of human prion disease. However, bulk expression analysis in two brain regions in sCJD displayed regionally distinct inflammatory profiles
[83][114].
Prion disease-associated astrogliosis is also prominent in human prion diseases as shown by the upregulation of GFAP and YKL-40 (or Chitinase-3-like protein 1; CHI3L1)
[60][84][73,107]. However, while a GFAP increase is also found in other neurodegenerative diseases such as AD
[78][97], YKL-40 upregulation is more specific for human CJD and might serve as a diagnostic marker
[84][107]. The microglia signature of sCJD patients differ from those of rapid AD, with significant downregulation of the microglia homeostatic marker TMEM119
[78][97].
The regional differences in inflammation-associated expression changes are also beginning to be investigated and described in the frontal cortex and cerebellum of patients with sCJD
[83][114]. Neuronal loss shows considerable variation between various regions of the brain within a CJD-diseased individual and between CJD patients. However, the cortex areas and cerebellum are often severely affected in sCJD, with a relative sparing of the hippocampus and subcortical grey matter
[85][86][117,118]. Thus, it will be of the highest interest to investigate, if other, less-affected brain regions will show distinct inflammatory profiles in human prion diseases. Although the deposition patterns of misfolded PrP
Sc are diverse in murine models of prion disease, the heterogeneity of PrP
Sc deposition patterns is higher in human prion diseases and comprises synaptic, perivacuolar, plaque-like, kuru-plaque, florid-plaque, punctuate, perineuronal, and intraneuronal patterns, which might occur side-by-side or even overlapping in the same patient
[87][88][119,120]. If and how these protein deposits directly influence the inflammatory profiles in individual brain regions in human prion diseases is currently not clear.