 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Joseph AMJL Janssen | -- | 2508 | 2023-03-02 16:56:12 | | | |
| 2 | Joseph AMJL Janssen | + 16 word(s) | 2524 | 2023-03-02 23:58:57 | | | | |
| 3 | Peter Tang | + 1 word(s) | 2525 | 2023-03-03 02:17:10 | | | | |
| 4 | Peter Tang | Meta information modification | 2525 | 2023-03-03 02:17:35 | | |
Video Upload Options
The metabolic syndrome is a cluster of overlapping conditions resulting in an increased incidence of type 2 diabetes, cardiovascular disease, and cancer. In the last few decades, prevalence of the metabolic syndrome in the Western world has reached epidemic proportions and this is likely due to alterations in diet and the environment as well as decreased physical activity. The Western diet and lifestyle (Westernization) plays an important etiological role in the pathogenesis of the metabolic syndrome by exerting negative effects on activity of the insulin–insulin-like growth factor-I (insulin–IGF-I) system. Interventions that normalize/reduce activity of the insulin–IGF-I system may play a key role in the prevention and treatment of the metabolic syndrome. For successful prevention, limitation, and treatment of the metabolic syndrome, the focus should be primarily on changing our diets and lifestyle in accordance with our genetic make-up, formed in adaptation to Paleolithic diets and lifestyles during a period of several million years of human evolution.
1. The Insulin–IGF-I System
2. Insulin Resistance, Hyperinsulinemia and the Metabolic Syndrome
3. Effects of Hyperinsulinemia on the Balance of the Insulin–GH–IGF-I Axis
4. Low(er) Activity of Insulin/IGF-I Signaling Pathway Protects against Type 2 Diabetes and Cancer
5. The Activity of the Insulin–IGF-I Signaling Pathway and Longevity
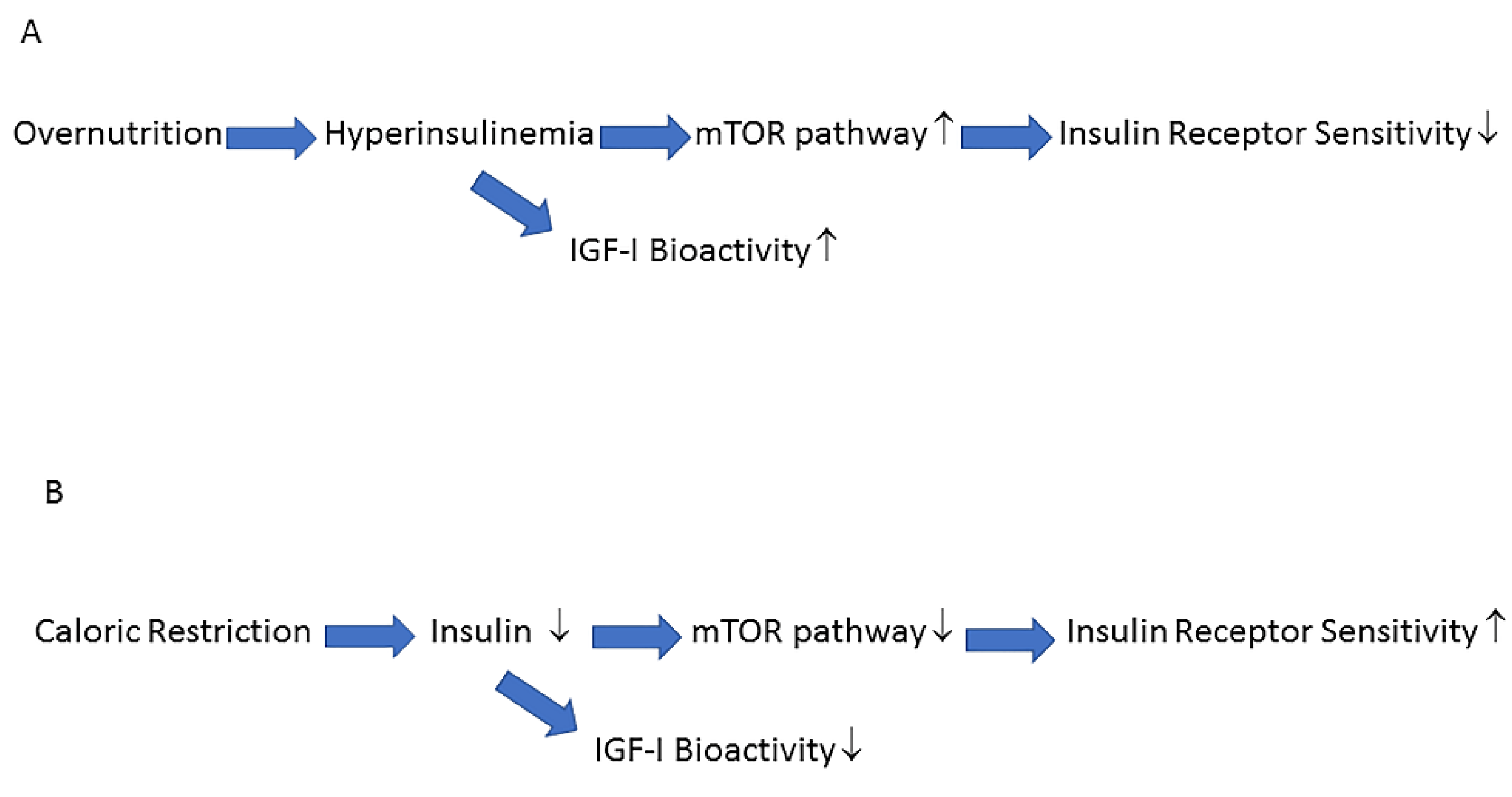
6. How to Halt the Negative Impact of the Western Lifestyle on the Insulin/IGF-I System and the Prevalence of the Metabolic Syndrome
References
- Collett-Solberg, P.F.; Cohen, P. The role of the insulin-like growth factor binding proteins and the igfbp proteases in modulating igf action. Endocrinol. Metab. Clin. N. Am. 1996, 25, 591–614.
- Clemmons, D.R. Role of IGF-binding proteins in regulating IGF responses to changes in metabolism. J. Mol. Endocrinol. 2018, 61, T139–T169.
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin Receptor Isoforms and Insulin Receptor/Insulin-Like Growth Factor Receptor Hybrids in Physiology and Disease. Endocr. Rev. 2009, 30, 586–623.
- Scalia, P.; Williams, S.J.; Fujita-Yamaguchi, Y.; Giordano, A. Cell cycle control by the insulin-like growth factor signal: At the crossroad between cell growth and mitotic regulation. Cell Cycle 2023, 22, 1–37.
- Scalia, P.; Giordano, A.; Williams, S.J. The IGF-II–Insulin Receptor Isoform-A Autocrine Signal in Cancer: Actionable Perspectives. Cancers 2020, 12, 366.
- Rosenzweig, S.A. What’s new in the IGF-binding proteins? Growth Horm. IGF Res. 2004, 14, 329–336.
- Chan, S.J.; Cao, Q.P.; Steiner, D.F. Evolution of the insulin superfamily: Cloning of a hybrid insulin/insulin-like growth factor cDNA from amphioxus. Proc. Natl. Acad. Sci. USA 1990, 87, 9319–9323.
- Chan, S.; Steiner, D.F. Insulin through the ages: Phylogeny of a growth promoting and metabolic regulatory hormone. Am. Zool. 2000, 40, 213–222.
- Taguchi, A.; White, M.F. Insulin-Like Signaling, Nutrient Homeostasis, and Life Span. Annu. Rev. Physiol. 2008, 70, 191–212.
- Cornier, M.-A.; Dabelea, D.; Hernandez, T.L.; Lindstrom, R.C.; Steig, A.J.; Stob, N.R.; Van Pelt, R.E.;Wang, H.; Eckel, R.H. The Metabolic Syndrome. Endocr. Rev. 2008, 29, 777–822
- Liese, A.D.; Mayer-Davis, E.J.; Tyroler, H.A.; Davis, C.E.; Keil, U.; Duncan, B.B.; Heiss, G. Development of the multiple metabolic syndrome in the ARIC cohort: Joint contribution of insulin, BMI, and WHR. Atherosclerosis risk in communities. Ann. Epidemiol.1997, 7, 407–416
- Liese, A.D.; Mayer-Davis, E.J.; Tyroler, H.A.; Davis, C.E.; Keil, U.; Duncan, B.B.; Heiss, G. Development of the multiple metabolic syndrome in the ARIC cohort: Joint contribution of insulin, BMI, and WHR. Atherosclerosis risk in communities. Ann. Epidemiol. 1997, 7, 407–416
- Hoppe, C.; Mølgaard, C.; Vaag, A.; Barkholt, V.; Michaelsen, K.F. High intakes of milk, but not meat, increase s-insulin and insulin resistance in 8-year-old boys. Eur. J. Clin. Nutr. 2005, 59, 393–398.
- Le Stunff, C.; Bougnères, P. Early Changes in Postprandial Insulin Secretion, Not in Insulin Sensitivity, Characterize Juvenile Obesity. Diabetes 1994, 43, 696–702
- Ng, Y.; Ramm, G.; James, D.E. Dissecting the Mechanism of Insulin Resistance Using a Novel Heterodimerization Strategy to Activate Akt. J. Biol. Chem. 2010, 285, 5232–5239
- Rizza, R.A.; Mandarino, L.J.; Genest, J.; Baker, B.A.; Gerich, J.E. Production of insulin resistance by hyperinsulinaemia in man. Diabetologia 1985, 28, 70–75.
- Del Prato, S.; Leonetti, F.; Simonson, D.C.; Sheehan, P.; Matsuda, M.; DeFronzo, R.A. Effect of sustained physiologic hyperinsulinaemia and hyperglycaemia on insulin secretion and insulin sensitivity in man. Diabetologia 1994, 37, 1025–1035.
- Weyer, C.; Hanson, R.L.; Tataranni, P.A.; Bogardus, C.; Pratley, R.E. A high fasting plasma insulin concentration predicts type 2 diabetes independent of insulin resistance: Evidence for a pathogenic role of relative hyperinsulinemia. Diabetes 2000, 49, 2094–2101.
- Dankner, R.; Chetrit, A.; Shanik, M.H.; Raz, I.; Roth, J. Basal state hyperinsulinemia in healthy normoglycemic adults heralds dysglycemia after more than two decades of follow up. Diabetes Metab. Res. Rev. 2012, 28, 618–624
- Tricò, D.; Natali, A.; Arslanian, S.; Mari, A.; Ferrannini, E. Identification, pathophysiology, and clinical implications of primary insulin hypersecretion in nondiabetic adults and adolescents. J. Clin. Investig. 2018, 3, e124912.
- Brøns, C.; Jensen, C.B.; Storgaard, H.; Hiscock, N.J.; White, A.; Appel, J.S.; Jacobsen, S.; Nilsson, E.; Larsen, C.M.; Astrup, A.; et al. Impact of short-term high-fat feeding on glucose and insulin metabolism in young healthy men. J. Physiol. 2009, 587, 2387–2397.
- Ferrannini, E.; Natali, A.; Bell, P.; Cavalloperin, P.; Lalic, N.; Mingrone, G. Insulin resistance and hypersecretion in obesity. European Group for the Study of Insulin Resistance (EGIR). J. Clin. Investig. 1997, 100, 1166–1173.
- Kim, M.K.; Reaven, G.M.; Kim, S.H. Dissecting the relationship between obesity and hyperinsulinemia: Role of insulin secretion and insulin clearance. Obesity 2016, 25, 378–383.
- Schofield, C.J.; Sutherland, C. Disordered insulin secretion in the development of insulin resistance and Type 2 diabetes. Diabet. Med. 2012, 29, 972–979.
- Shanik, M.H.; Xu, Y.; Škrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin Resistance and Hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care 2008, 31 (Suppl. 2), S262–S268.
- Corkey, B.E. Banting Lecture 2011: Hyperinsulinemia: Cause or Consequence? Diabetes 2011, 61, 4–13.
- Sung, K.-C.C.; Seo, M.-H.H.; Rhee, E.-J.J.; Wilson, A.M. Elevated fasting insulin predicts the future incidence of metabolic syndrome: A 5-year follow-up study. Cardiovasc. Diabetol. 2011, 10, 108.
- Mykkänen, L.; Kuusisto, J.; Haffner, S.M.; Pyörälä, K.; Laakso, M. Hyperinsulinemia predicts multiple atherogenic changes in lipoproteins in elderly subjects. Arter. Thromb. 1994, 14, 518–526.
- Charles, M.A.; Fontbonne, A.; Thibult, N.;Warnet, J.-M.; Rosselin, G.E.; Eschwege, E. Risk Factors for NIDDM in White Population: Paris Prospective Study. Diabetes 1991, 40, 796–799.
- Salonen, J.T.; Lakka, T.A.; Lakka, H.-M.; Valkonen, V.-P.; Everson, S.; Kaplan, G.A. Hyperinsulinemia Is Associated With the Incidence of Hypertension and Dyslipidemia in Middle-Aged Men. Diabetes 1998, 47, 270–275.
- Cusin, I.; Rohner-Jeanrenaud, F.; Terrettaz, J.; Jeanrenaud, B. Hyperinsulinemia and its impact on obesity and insulin resistance. Int. J. Obes. Relat. Metab. Disord. 1992, 16 (Suppl. 4), S1–S11.
- Simmons, A.L.; Schlezinger, J.J.; Corkey, B.E. What Are We Putting in Our Food That Is Making Us Fat? Food Additives, Contaminants, and Other Putative Contributors to Obesity. Curr. Obes. Rep. 2014, 3, 273–285.
- Bergman, R.N.; Piccinini, F.; Kabir, M.; Kolka, C.M.; Ader, M. Hypothesis: Role of Reduced Hepatic Insulin Clearance in the Pathogenesis of Type 2 Diabetes. Diabetes 2019, 68, 1709–1716.
- Hartman, M.L.; E Clayton, P.; Johnson, M.L.; Celniker, A.; Perlman, A.J.; Alberti, K.G.; Thorner, M.O. A low dose euglycemic infusion of recombinant human insulin-like growth factor I rapidly suppresses fasting-enhanced pulsatile growth hormone secretion in humans. J. Clin. Investig. 1993, 91, 2453–2462.
- Janssen, J. Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer. Int. J. Mol. Sci. 2021, 22, 7797.
- Frystyk, J.; Vestbo, E.; Skjærbaek, C.; Mogensen, C.; Ørskov, H. Free insulin-like growth factors in human obesity. Metabolism 1995, 44, 37–44.
- Brugts, M.P.; van Duijn, C.M.; Hofland, L.J.;Witteman, J.C.; Lamberts, S.W.; Janssen, J.A. IGF-I Bioactivity in an Elderly Population: Relation to insulin sensitivity, insulin levels, and the metabolic syndrome. Diabetes 2010, 59, 505–508.
- Laron, Z. Laron Syndrome (Primary Growth Hormone Resistance or Insensitivity): The Personal Experience 1958–2003. J. Clin. Endocrinol. Metab. 2004, 89, 1031–1044.
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Cheng, C.-W.; Hwang, D.; Martin-Montalvo, A.; Saavedra, J.; Ingles, S.; et al. Growth Hormone Receptor Deficiency Is Associated with a Major Reduction in Pro-Aging Signaling, Cancer, and Diabetes in Humans. Sci. Transl. Med. 2011, 3, 70ra13.
- Melnik, B.C.; John, S.M.; Schmitz, G. Over-stimulation of insulin/IGF-1 signaling by western diet may promote diseases of civilization: Lessons learnt from laron syndrome. Nutr. Metab. 2011, 8, 41.
- Barbieri, M.; Bonafè, M.; Franceschi, C.; Paolisso, G. Insulin/IGF-I-signaling pathway: An evolutionarily conserved mechanism of longevity from yeast to humans. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E1064–E1071.
- Brown-Borg, H.M. Hormonal regulation of aging and life span. Trends Endocrinol. Metab. 2003, 14, 151–153.
- Longo, V.D.; Finch, C.E. Evolutionary Medicine: From Dwarf Model Systems to Healthy Centenarians? Science 2003, 299, 1342–1346.
- Fontana, L.; Partridge, L.; Longo, V.D. Extending Healthy Life Span—From Yeast to Humans. Science 2010, 328, 321–326.
- Vitale, G.; Brugts, M.P.; Ogliari, G.; Castaldi, D.; Fatti, L.M.; Varewijck, A.J.; Lamberts, S.W.; Monti, D.; Bucci, L.; Cevenini, E.; et al. Low circulating IGF-I bioactivity is associated with human longevity: Findings in centenarians’ offspring. Aging 2012, 4, 580–589.
- Blagosklonny, M.V. Calorie restriction: Decelerating mTOR-driven aging from cells to organisms (including humans). Cell Cycle 2010, 9, 683–688.
- Blagosklonny, M.V. Once again on rapamycin-induced insulin resistance and longevity: Despite of or owing to. Aging 2012, 4, 350–358.
- Parr, T. Insulin exposure controls the rate of mammalian aging. Mech. Ageing Dev. 1996, 88, 75–82.
- Paolisso, G.; Gambardella, A.; Ammendola, S.; D’Amore, A.; Balbi, V.; Varricchio, M.; D’Onofrio, F. Glucose tolerance and insulin action in healthy centenarians. Am. J. Physiol. Metab. 1996, 270, E890–E894.
- Barbieri, M.; Rizzo, M.R.; Manzella, D.; Paolisso, G. Age-related insulin resistance: Is it an obligatory finding? The lesson from healthy centenarians. Diabetes Metab. Res. Rev. 2001, 17, 19–26.
- Paolisso, G.; Tagliamonte, M.R.; Rizzo, M.R.; Manzella, D.; Gambardella, A.; Varricchio, M. Oxidative Stress and Advancing Age: Results in Healthy Centenarians. J. Am. Geriatr. Soc. 1998, 46, 833–838.
- Matsuzawa-Nagata, N.; Takamura, T.; Ando, H.; Nakamura, S.; Kurita, S.; Misu, H.; Ota, T.; Yokoyama, M.; Honda, M.; Miyamoto, K.-I.; et al. Increased oxidative stress precedes the onset of high-fat diet–induced insulin resistance and obesity. Metabolism 2008, 57, 1071–1077.
- Kopp, W. How Western Diet And Lifestyle Drive The Pandemic Of Obesity And Civilization Diseases. Diabetes Metab. Syndr. Obes. 2019, 12, 2221–2236.
- Kopp,W. Diet-Induced Hyperinsulinemia as a Key Factor in the Etiology of Both Benign Prostatic Hyperplasia and Essential Hypertension? Nutr. Metab. Insights 2018, 11, 1178638818773072.
- Esmaillzadeh, A.; Kimiagar, M.; Mehrabi, Y.; Azadbakht, L.; Hu, F.B.; Willett, W.C. Dietary patterns, insulin resistance, and prevalence of the metabolic syndrome in women. Am. J. Clin. Nutr. 2007, 85, 910–918. [
- Eaton, S.B.; Konner, M. Paleolithic nutrition. A consideration of its nature and current implications. N. Engl. J. Med. 1985, 312, 283–289.
- O’Keefe, J.H.; Cordain, L. Cardiovascular Disease Resulting from a Diet and Lifestyle at Odds With Our Paleolithic Genome: How to Become a 21st-Century Hunter-Gatherer. Mayo Clin. Proc. 2004, 79, 101–108.


