The metabolic syndrome is a cluster of overlapping conditions resulting in an increased incidence of type 2 diabetes, cardiovascular disease, and cancer. In the last few decades, prevalence of the metabolic syndrome in the Western world has reached epidemic proportions and this is likely due to alterations in diet and the environment as well as decreased physical activity. The Western diet and lifestyle (Westernization) plays an important etiological role in the pathogenesis of the metabolic syndrome by exerting negative effects on activity of the insulin–insulin-like growth factor-I (insulin–IGF-I) system. Interventions that normalize/reduce activity of the insulin–IGF-I system may play a key role in the prevention and treatment of the metabolic syndrome. For successful prevention, limitation, and treatment of the metabolic syndrome, the focus should be primarily on changing our diets and lifestyle in accordance with our genetic make-up, formed in adaptation to Paleolithic diets and lifestyles during a period of several million years of human evolution.
- hyperinsulinemia
- IGF-I
- insulin resistance
- IGFBP-1
- Metabolic Syndrome
- Westernization
- Overnutrition
- Paleloithic diet
- Meditererraenean diet
- Prevention
- Paleoithic diet
- Meditererranean diet
1. The Insulin–IGF-I System
2. Insulin Resistance, Hyperinsulinemia and the Metabolic Syndrome
appear to be central in the development of the metabolic syndrome [10][12]. In the view of Reaven (see above) and in most traditional literature, obesity is considered
the main cause of insulin resistance, and insulin resistance is the abnormality leading to hyperinsulinemia. In this model, hyperinsulinemia is a compensatory response to insulin
resistance. However, an increasing number of (prospective) human studies suggests an alternative scenario: in this scenario, chronic hypersecretion of insulin is the primary abnormality leading to hyperinsulinemia and precedes, initiates, and causes insulin resistance. In this new scenario, hyperinsulinemia is the first event triggering insulin resistance, obesity, the metabolic syndrome, and type 2 diabetes [13][14][15][16][17][18][19][20][21][22][23]. In addition, in subjects with normal plasma glucose concentrations, it has been found that hyperinsulinemia per se induced insulin resistance by insulin-induced downregulation of insulin receptor signaling [24][25]. This further supports the idea that hyperinsulinemia may be a primary driver of insulin resistance [25][26].
future development of the metabolic syndrome [27]. In addition, prospective evidence shows that—independently of obesity and body weight—hyperinsulinemia is related to
the development of dyslipidemia and hypertension, suggesting that hyperinsulinemia precedes these disorders in the etiologic pathway [28][29][30]. Thus, hyperinsulinemia may
indeed be an early and central feature of the cardiovascular risk of subjects with the metabolic syndrome [27]. Further support of a pathogenic role of hyperinsulinemia in
the development of the metabolic syndrome was demonstrated in an animal model by Jeanrenaud et al. They showed that short-term hyperinsulinemia is a pathological driving
force, which produces incipient obesity by overstimulating white adipose tissue and liver metabolic activity while concomitantly producing incipient muscle insulin resistance [31].
in turn exacerbates insulin resistance [33]. Consequently, insulin resistance will stress pancreatic beta-cells, and this may finally result in their ultimate failure and onset of frank type 2 diabetes. In addition, development of insulin resistance in muscles and fat cells is caused by of an overexposure of the (post-hepatic) peripheral tissues to endogenous insulin [33]. As a direct consequence of the peripheral hyperinsulinemia, peripheral insulin resistance develops to dampen hyperinsulinemia-mediated stimulating effects in muscles and fat cells [33].
3. Effects of Hyperinsulinemia on the Balance of the Insulin–GH–IGF-I Axis
liver feeds back to suppress both insulin and GH [34][35]. The typical modern Western diet can disturb this balance. Due to its continuous food intake, energy surplus,
high content of sugars, corn-derived fructose syrup, saturated fats and proteins, the modern Western diet may induce hyperinsulinemia, which in turn increases IGF-I secretion [35].
Increased IGF-I subsequently induces suppression of GH secretion to lower levels than normal [36][37]. Only a few days of overeating may markedly suppress GH secretion before any measurable weight gain, and it has been suggested that, in these circumstances, the accompanying hyperinsulinemia is a likely mediator of this rapid reduction in GH secretion. Consequently, a shift of the insulin : GH ratio towards insulin (and IGF-I) and away from GH will occur. The higher insulin : GH ratio lowers energy expenditure and induces fat accumulation, thereby promoting energy storage and lipid synthesis and hindering lipid breakdown. This will promote obesity because of higher fat accumulation and lower energy expenditure.
4. Low(er) Activity of Insulin/IGF-I Signaling Pathway Protects against Type 2 Diabetes and Cancer
Laron syndrome is a disorder characterized by a lack of IGF-I production in response to GH [38]. It is caused by inherited GH receptor mutations and Individuals with the classic Laron syndrome present with short stature, obesity, low blood sugar, and congenital IGF-I deficiency (with low serum IGF-I) with decreased insulin/IGF-I signaling activity despite elevated basal serum GH [38]. Guevera-Aguire et al. found in individuals with Laron syndrome, in contrast to their healthy relatives with normal insulin/IGF-I signaling, a significant reduction in pro-aging signaling, cancer, and type 2 diabetes [39]. Serum from subjects with the Laron syndrome induced in vitro a reduced number of DNA breaks but increased apoptosis in human mammary epithelial cells treated by hydrogen peroxide [39]. Moreover, serum from subjects with the Laron syndrome also caused reduced expression of rat sarcoma virus (RAS), PKA (protein kinase A), and mTOR (target of rapamycin) and up-regulation of superoxide dismutase 2 in treated cells [39]. All these changes promote normal cellular protection and life-span extension in model organisms and provide a possible explanation for the observed low incidence of cancer in subjects with the Laron syndrome in the study by Guevera-Aguire et al. [39]. Moreover, individuals with the Laron syndrome showed reduced insulin concentrations (1.4 μU/mL versus 4.4 μU/mL in unaffected relatives) and a very low HOMA-IR (homeostatic model assessment-insulin resistance) index (0.34 versus 0.96 in unaffected relatives), indicating that higher insulin sensitivity could provide a possible (alternative) mechanism explaining the reduced prevalence of type 2 diabetes and cancer observed in subjects with the Laron syndrome [39]. Thus Laron syndrome, an experiment of nature, showed that in humans, low(er) activity of the insulin/IGF-I signaling pathway and high insulin sensitivity may play a crucial role in the protection from diseases typically related to Western civilization, such as type 2 diabetes and cancer [40].4. The Activity of the Insulin–IGF-I Signaling Pathway and Longevity
Disruption of genes in the insulin–IGF-I signaling pathway that share similarities with those in humans can significantly extend life span in diverse species, including yeast, worms, fruit flies, and rodents [41]. It has therefore been suggested that reducing the activity of the insulin–IGF-I signaling pathway plays a key role in delayed aging and prolonged longevity [42]. A point to emphasize here, and in favor of this suggestion, is that all long-lived mutants, ranging from yeast to mice, share some important phenotypic characteristics, including reduced insulin signaling, enhanced insulin sensitivity, and reduced IGF-I plasma levels [41]. Laboratory animals fed ad libitum have relatively low levels of physical activity and therefore show similarities to humans with a Western (sedentary) lifestyle, who are at high risk for hyperinsulinemia, obesity, and insulin resistance [43]. Caloric restriction counteracts the general trend for laboratory animals to progressively increase fat mass during aging [43]. Restriction of the number of calories consumed lowers the incidence of age-related loss of functions and disease and increases life span in a wide variety of animals [43][44]. It has been further suggested that many of the beneficial effects of caloric restriction on lifespan are mediated, at least in part, by down-regulation of the insulin/IGF-I signaling pathway activity [45]. In support of this latter option is the observation that caloric restriction in rodents decreases activity of the insulin/IGF-I signaling cascade and postpones or attenuates cancer, immunosenescence, and inflammation without irreversible side effects [43]. Nutrients and insulin both activate the mTOR pathway and this pathway is involved in cellular senescence and age-related diseases [45][46][47]. Overnutrition increases insulin secretion, increases IGF-I bioactivity and hyperactivates the mTOR pathway [47] (Figure 1A). Hyperactivation of the mTOR pathway induces insulin receptor resistance which blocks insulin-mediated glucose uptake and results in elevated glucose levels [47]. The elevated glucose levels induce a further increase in insulin secretion, which will, in turn, further deteriorate insulin sensitivity [45]. In contrast, lifespan-extending caloric restriction without malnutrition, decreases the activity of the mTOR pathway: this improves insulin receptor sensitivity and secondarily reduces plasma glucose levels, insulin levels and IGF-I bioactivity [45][46] (Figure 1B).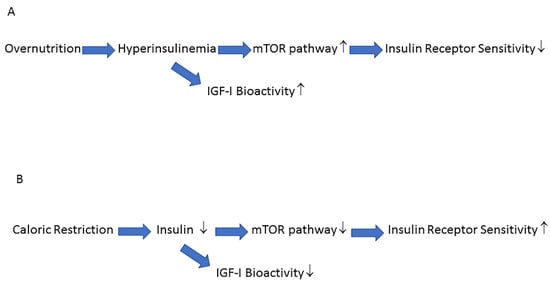
5. How to Halt the Negative Impact of the Western Lifestyle on the Insulin/IGF-I System and the Prevalence of the Metabolic Syndrome
The increasing prevalence of the metabolic syndrome seems primarily driven by changes in diet and increasingly sedentary lifestyles [53]. Due to the Western dietarypattern of frequent snacking and frequent consumption of sucrose-containing soft drinks, insulin levels are elevated most of the day [54][55]. The changes in dietary habits, adopted by the Western world over the past 100 years, appear to have made an important contribution to the increasing prevalence of the metabolic syndrome and its consequences—coronary artery disease, hypertension, diabetes, and some cancers. These conditions have only emerged in the past century but were virtually absent in hunter-gatherer populations following a traditional hunter-gatherer (or paleolithic) diet and lifestyle [56]. However, genetic make-up of humans is still, at present, best adapted to the low-glycemic and low-insulinemic hunter-gatherer (paleolitic) diet, and it therefore has been suggested that, although we are people living in the 21st century, genetically we are still citizens of the
Paleolithic era [54][57]. As discussed previously, the “modern” Western diet and lifestyle, by exerting negative (i.e., stimulating) effects on the activity of the insulin–IGF-I system, may play an
important etiological role in the pathogenesis of the metabolic syndrome. Interventions that normalize/reduce activity of the insulin–IGF-I system might therefore play a key role
in the prevention and treatment of the metabolic syndrome and its consequences. Primary prevention of the metabolic syndrome (i.e., before it starts) is probably the only effective and cost-effective approach, counterbalancing the environmental roots of the
increased prevalence of the metabolic syndrome. Some advocate that we should turn to the paleolithic diet and lifestyle when seeking solutions for the increased prevalence of
hyperinsulinemia, insulin resistance, obesity, and type 2 diabetes, which have emerged in many populations worldwide after switching to the Westernized way of life [56]. Modifications of our diets and lifestyle, in accordance with our genetic constitution-formed in adaptation to Paleolithic diets and lifestyles during a period of several million
years of human evolution—may help prevent or limit the development of the metabolic syndrome. Translating this insight into clinical practice, however, requires not only individual changes in our food and lifestyle and the early start and adoption of healthy habits at a young age in pediatric populations, but also requires fundamental changes
in our health system and food industry.
References
- Collett-Solberg, P.F.; Cohen, P. The role of the insulin-like growth factor binding proteins and the igfbp proteases in modulating igf action. Endocrinol. Metab. Clin. N. Am. 1996, 25, 591–614.
- Clemmons, D.R. Role of IGF-binding proteins in regulating IGF responses to changes in metabolism. J. Mol. Endocrinol. 2018, 61, T139–T169.
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin Receptor Isoforms and Insulin Receptor/Insulin-Like Growth Factor Receptor Hybrids in Physiology and Disease. Endocr. Rev. 2009, 30, 586–623.
- Scalia, P.; Williams, S.J.; Fujita-Yamaguchi, Y.; Giordano, A. Cell cycle control by the insulin-like growth factor signal: At the crossroad between cell growth and mitotic regulation. Cell Cycle 2023, 22, 1–37.
- Scalia, P.; Giordano, A.; Williams, S.J. The IGF-II–Insulin Receptor Isoform-A Autocrine Signal in Cancer: Actionable Perspectives. Cancers 2020, 12, 366.
- Rosenzweig, S.A. What’s new in the IGF-binding proteins? Growth Horm. IGF Res. 2004, 14, 329–336.
- Chan, S.J.; Cao, Q.P.; Steiner, D.F. Evolution of the insulin superfamily: Cloning of a hybrid insulin/insulin-like growth factor cDNA from amphioxus. Proc. Natl. Acad. Sci. USA 1990, 87, 9319–9323.
- Chan, S.; Steiner, D.F. Insulin through the ages: Phylogeny of a growth promoting and metabolic regulatory hormone. Am. Zool. 2000, 40, 213–222.
- Taguchi, A.; White, M.F. Insulin-Like Signaling, Nutrient Homeostasis, and Life Span. Annu. Rev. Physiol. 2008, 70, 191–212.
- Cornier, M.-A.; Dabelea, D.; Hernandez, T.L.; Lindstrom, R.C.; Steig, A.J.; Stob, N.R.; Van Pelt, R.E.;Wang, H.; Eckel, R.H. The Metabolic Syndrome. Endocr. Rev. 2008, 29, 777–822
- Liese, A.D.; Mayer-Davis, E.J.; Tyroler, H.A.; Davis, C.E.; Keil, U.; Duncan, B.B.; Heiss, G. Development of the multiple metabolic syndrome in the ARIC cohort: Joint contribution of insulin, BMI, and WHR. Atherosclerosis risk in communities. Ann. Epidemiol.1997, 7, 407–416
- Liese, A.D.; Mayer-Davis, E.J.; Tyroler, H.A.; Davis, C.E.; Keil, U.; Duncan, B.B.; Heiss, G. Development of the multiple metabolic syndrome in the ARIC cohort: Joint contribution of insulin, BMI, and WHR. Atherosclerosis risk in communities. Ann. Epidemiol. 1997, 7, 407–416
- Hoppe, C.; Mølgaard, C.; Vaag, A.; Barkholt, V.; Michaelsen, K.F. High intakes of milk, but not meat, increase s-insulin and insulin resistance in 8-year-old boys. Eur. J. Clin. Nutr. 2005, 59, 393–398.
- Le Stunff, C.; Bougnères, P. Early Changes in Postprandial Insulin Secretion, Not in Insulin Sensitivity, Characterize Juvenile Obesity. Diabetes 1994, 43, 696–702
- Ng, Y.; Ramm, G.; James, D.E. Dissecting the Mechanism of Insulin Resistance Using a Novel Heterodimerization Strategy to Activate Akt. J. Biol. Chem. 2010, 285, 5232–5239
- Rizza, R.A.; Mandarino, L.J.; Genest, J.; Baker, B.A.; Gerich, J.E. Production of insulin resistance by hyperinsulinaemia in man. Diabetologia 1985, 28, 70–75.
- Del Prato, S.; Leonetti, F.; Simonson, D.C.; Sheehan, P.; Matsuda, M.; DeFronzo, R.A. Effect of sustained physiologic hyperinsulinaemia and hyperglycaemia on insulin secretion and insulin sensitivity in man. Diabetologia 1994, 37, 1025–1035.
- Weyer, C.; Hanson, R.L.; Tataranni, P.A.; Bogardus, C.; Pratley, R.E. A high fasting plasma insulin concentration predicts type 2 diabetes independent of insulin resistance: Evidence for a pathogenic role of relative hyperinsulinemia. Diabetes 2000, 49, 2094–2101.
- Dankner, R.; Chetrit, A.; Shanik, M.H.; Raz, I.; Roth, J. Basal state hyperinsulinemia in healthy normoglycemic adults heralds dysglycemia after more than two decades of follow up. Diabetes Metab. Res. Rev. 2012, 28, 618–624
- Tricò, D.; Natali, A.; Arslanian, S.; Mari, A.; Ferrannini, E. Identification, pathophysiology, and clinical implications of primary insulin hypersecretion in nondiabetic adults and adolescents. J. Clin. Investig. 2018, 3, e124912.
- Brøns, C.; Jensen, C.B.; Storgaard, H.; Hiscock, N.J.; White, A.; Appel, J.S.; Jacobsen, S.; Nilsson, E.; Larsen, C.M.; Astrup, A.; et al. Impact of short-term high-fat feeding on glucose and insulin metabolism in young healthy men. J. Physiol. 2009, 587, 2387–2397.
- Ferrannini, E.; Natali, A.; Bell, P.; Cavalloperin, P.; Lalic, N.; Mingrone, G. Insulin resistance and hypersecretion in obesity. European Group for the Study of Insulin Resistance (EGIR). J. Clin. Investig. 1997, 100, 1166–1173.
- Kim, M.K.; Reaven, G.M.; Kim, S.H. Dissecting the relationship between obesity and hyperinsulinemia: Role of insulin secretion and insulin clearance. Obesity 2016, 25, 378–383.
- Schofield, C.J.; Sutherland, C. Disordered insulin secretion in the development of insulin resistance and Type 2 diabetes. Diabet. Med. 2012, 29, 972–979.
- Shanik, M.H.; Xu, Y.; Škrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin Resistance and Hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care 2008, 31 (Suppl. 2), S262–S268.
- Corkey, B.E. Banting Lecture 2011: Hyperinsulinemia: Cause or Consequence? Diabetes 2011, 61, 4–13.
- Sung, K.-C.C.; Seo, M.-H.H.; Rhee, E.-J.J.; Wilson, A.M. Elevated fasting insulin predicts the future incidence of metabolic syndrome: A 5-year follow-up study. Cardiovasc. Diabetol. 2011, 10, 108.
- Mykkänen, L.; Kuusisto, J.; Haffner, S.M.; Pyörälä, K.; Laakso, M. Hyperinsulinemia predicts multiple atherogenic changes in lipoproteins in elderly subjects. Arter. Thromb. 1994, 14, 518–526.
- Charles, M.A.; Fontbonne, A.; Thibult, N.;Warnet, J.-M.; Rosselin, G.E.; Eschwege, E. Risk Factors for NIDDM in White Population: Paris Prospective Study. Diabetes 1991, 40, 796–799.
- Salonen, J.T.; Lakka, T.A.; Lakka, H.-M.; Valkonen, V.-P.; Everson, S.; Kaplan, G.A. Hyperinsulinemia Is Associated With the Incidence of Hypertension and Dyslipidemia in Middle-Aged Men. Diabetes 1998, 47, 270–275.
- Cusin, I.; Rohner-Jeanrenaud, F.; Terrettaz, J.; Jeanrenaud, B. Hyperinsulinemia and its impact on obesity and insulin resistance. Int. J. Obes. Relat. Metab. Disord. 1992, 16 (Suppl. 4), S1–S11.
- Simmons, A.L.; Schlezinger, J.J.; Corkey, B.E. What Are We Putting in Our Food That Is Making Us Fat? Food Additives, Contaminants, and Other Putative Contributors to Obesity. Curr. Obes. Rep. 2014, 3, 273–285.
- Bergman, R.N.; Piccinini, F.; Kabir, M.; Kolka, C.M.; Ader, M. Hypothesis: Role of Reduced Hepatic Insulin Clearance in the Pathogenesis of Type 2 Diabetes. Diabetes 2019, 68, 1709–1716.
- Hartman, M.L.; E Clayton, P.; Johnson, M.L.; Celniker, A.; Perlman, A.J.; Alberti, K.G.; Thorner, M.O. A low dose euglycemic infusion of recombinant human insulin-like growth factor I rapidly suppresses fasting-enhanced pulsatile growth hormone secretion in humans. J. Clin. Investig. 1993, 91, 2453–2462.
- Janssen, J. Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer. Int. J. Mol. Sci. 2021, 22, 7797.
- Frystyk, J.; Vestbo, E.; Skjærbaek, C.; Mogensen, C.; Ørskov, H. Free insulin-like growth factors in human obesity. Metabolism 1995, 44, 37–44.
- Brugts, M.P.; van Duijn, C.M.; Hofland, L.J.;Witteman, J.C.; Lamberts, S.W.; Janssen, J.A. IGF-I Bioactivity in an Elderly Population: Relation to insulin sensitivity, insulin levels, and the metabolic syndrome. Diabetes 2010, 59, 505–508.
- Laron, Z. Laron Syndrome (Primary Growth Hormone Resistance or Insensitivity): The Personal Experience 1958–2003. J. Clin. Endocrinol. Metab. 2004, 89, 1031–1044.
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Cheng, C.-W.; Hwang, D.; Martin-Montalvo, A.; Saavedra, J.; Ingles, S.; et al. Growth Hormone Receptor Deficiency Is Associated with a Major Reduction in Pro-Aging Signaling, Cancer, and Diabetes in Humans. Sci. Transl. Med. 2011, 3, 70ra13.
- Melnik, B.C.; John, S.M.; Schmitz, G. Over-stimulation of insulin/IGF-1 signaling by western diet may promote diseases of civilization: Lessons learnt from laron syndrome. Nutr. Metab. 2011, 8, 41.
- Barbieri, M.; Bonafè, M.; Franceschi, C.; Paolisso, G. Insulin/IGF-I-signaling pathway: An evolutionarily conserved mechanism of longevity from yeast to humans. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E1064–E1071.
- Brown-Borg, H.M. Hormonal regulation of aging and life span. Trends Endocrinol. Metab. 2003, 14, 151–153.
- Longo, V.D.; Finch, C.E. Evolutionary Medicine: From Dwarf Model Systems to Healthy Centenarians? Science 2003, 299, 1342–1346.
- Fontana, L.; Partridge, L.; Longo, V.D. Extending Healthy Life Span—From Yeast to Humans. Science 2010, 328, 321–326.
- Vitale, G.; Brugts, M.P.; Ogliari, G.; Castaldi, D.; Fatti, L.M.; Varewijck, A.J.; Lamberts, S.W.; Monti, D.; Bucci, L.; Cevenini, E.; et al. Low circulating IGF-I bioactivity is associated with human longevity: Findings in centenarians’ offspring. Aging 2012, 4, 580–589.
- Blagosklonny, M.V. Calorie restriction: Decelerating mTOR-driven aging from cells to organisms (including humans). Cell Cycle 2010, 9, 683–688.
- Blagosklonny, M.V. Once again on rapamycin-induced insulin resistance and longevity: Despite of or owing to. Aging 2012, 4, 350–358.
- Parr, T. Insulin exposure controls the rate of mammalian aging. Mech. Ageing Dev. 1996, 88, 75–82.
- Paolisso, G.; Gambardella, A.; Ammendola, S.; D’Amore, A.; Balbi, V.; Varricchio, M.; D’Onofrio, F. Glucose tolerance and insulin action in healthy centenarians. Am. J. Physiol. Metab. 1996, 270, E890–E894.
- Barbieri, M.; Rizzo, M.R.; Manzella, D.; Paolisso, G. Age-related insulin resistance: Is it an obligatory finding? The lesson from healthy centenarians. Diabetes Metab. Res. Rev. 2001, 17, 19–26.
- Paolisso, G.; Tagliamonte, M.R.; Rizzo, M.R.; Manzella, D.; Gambardella, A.; Varricchio, M. Oxidative Stress and Advancing Age: Results in Healthy Centenarians. J. Am. Geriatr. Soc. 1998, 46, 833–838.
- Matsuzawa-Nagata, N.; Takamura, T.; Ando, H.; Nakamura, S.; Kurita, S.; Misu, H.; Ota, T.; Yokoyama, M.; Honda, M.; Miyamoto, K.-I.; et al. Increased oxidative stress precedes the onset of high-fat diet–induced insulin resistance and obesity. Metabolism 2008, 57, 1071–1077.
- Kopp, W. How Western Diet And Lifestyle Drive The Pandemic Of Obesity And Civilization Diseases. Diabetes Metab. Syndr. Obes. 2019, 12, 2221–2236.
- Kopp,W. Diet-Induced Hyperinsulinemia as a Key Factor in the Etiology of Both Benign Prostatic Hyperplasia and Essential Hypertension? Nutr. Metab. Insights 2018, 11, 1178638818773072.
- Esmaillzadeh, A.; Kimiagar, M.; Mehrabi, Y.; Azadbakht, L.; Hu, F.B.; Willett, W.C. Dietary patterns, insulin resistance, and prevalence of the metabolic syndrome in women. Am. J. Clin. Nutr. 2007, 85, 910–918. [
- Eaton, S.B.; Konner, M. Paleolithic nutrition. A consideration of its nature and current implications. N. Engl. J. Med. 1985, 312, 283–289.
- O’Keefe, J.H.; Cordain, L. Cardiovascular Disease Resulting from a Diet and Lifestyle at Odds With Our Paleolithic Genome: How to Become a 21st-Century Hunter-Gatherer. Mayo Clin. Proc. 2004, 79, 101–108.