Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Giovanni Vitale | -- | 2700 | 2023-02-24 08:35:23 | | | |
| 2 | Rita Xu | -1 word(s) | 2699 | 2023-02-24 09:00:35 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Vitale, G.; Mattiaccio, A.; Conti, A.; Turco, L.; Seri, M.; Piscaglia, F.; Morelli, M.C. Genetics in Familial Intrahepatic Cholestasis. Encyclopedia. Available online: https://encyclopedia.pub/entry/41617 (accessed on 24 July 2026).
Vitale G, Mattiaccio A, Conti A, Turco L, Seri M, Piscaglia F, et al. Genetics in Familial Intrahepatic Cholestasis. Encyclopedia. Available at: https://encyclopedia.pub/entry/41617. Accessed July 24, 2026.
Vitale, Giovanni, Alessandro Mattiaccio, Amalia Conti, Laura Turco, Marco Seri, Fabio Piscaglia, Maria Cristina Morelli. "Genetics in Familial Intrahepatic Cholestasis" Encyclopedia, https://encyclopedia.pub/entry/41617 (accessed July 24, 2026).
Vitale, G., Mattiaccio, A., Conti, A., Turco, L., Seri, M., Piscaglia, F., & Morelli, M.C. (2023, February 24). Genetics in Familial Intrahepatic Cholestasis. In Encyclopedia. https://encyclopedia.pub/entry/41617
Vitale, Giovanni, et al. "Genetics in Familial Intrahepatic Cholestasis." Encyclopedia. Web. 24 February, 2023.
Copy Citation
The family of inherited intrahepatic cholestasis includes autosomal recessive cholestatic rare diseases of childhood involved in bile acids secretion or bile transport defects. Specific genetic pathways potentially cause many otherwise unexplained cholestasis or hepatobiliary tumours in a healthy liver. Next-generation sequencing and whole-exome sequencing have improved the diagnostic procedures of familial intrahepatic cholestasis (FIC), as well as the discovery of several genes responsible for FIC.
progressive familial intrahepatic cholestasis
Alagille syndrome
hepatobiliary cancers
1. Introduction
Progressive familial intrahepatic cholestasis (PFIC), Alagille syndrome, ductal plaque abnormalities such as Caroli’s syndrome, congenital hepatic fibrosis, metabolic diseases including citrine deficiency, and finally, bile acid synthesis defects belong all to hereditary cholestatic disorders.
PFIC is a group of autosomal recessive cholestatic diseases caused by defects in hepatobiliary transport proteins, affecting especially newborns and children; since PFIC often evolves in liver failure and/or liver cancer, it represents an indication for liver transplantation (LT). In addition, the genes responsible for PFIC can cause other non-progressive biliary disorders: low-phospholipid-associated cholelithiasis (LPAC), benign recurrent intrahepatic cholestasis (BRIC), drug-induced cholestasis (DIC) and intrahepatic cholestasis of pregnancy (ICP) [1].
Bile acids (BAs) are steroid-based molecules synthesized from cholesterol in the liver and stored in the gallbladder. BAs regulate glucose and lipid homeostasis and drug absorption, functioning as molecules able to control energy metabolism and its several trafficking pathways.
Recently, the focus has moved to the enterohepatic circulation of BAs and gut microbiota; the misexpression of specific BAs transporters, combined with dysbiosis and imbalance of some gut microbes, have a crucial role in the modulation of inflammation, immunity and metabolism, finally leading to malignant transformation and development of hepatobiliary cancers (HBCs) [2][3][4].
In the last years, the continuous advances in diagnostic methods as well as next-generation sequencing (NGS), whole-exome sequencing (WES) and whole-genome sequencing (WGS) led to the finding of new genes responsible for cholestatic diseases and a better understanding of the relationship between a broad spectrum of cholestatic disorders and the development of HBCs.
As matter of fact then, several genes involved in the development of inherited cholestasis are linked to the risk of HCC and CCA in pediatric and not pediatric populations: ABCB11, ABCB4, TJP2, FXR, MYO5B, SLC51B, SLC25A13, NOTCH2, JAG1, TGR5 and HNF1B [5][6][7].
Furthermore, mutations in genes responsible for cholestasis may be present in children and adults with non-progressive forms of cryptogenic cholestasis, even in their heterozygous status [1][8].
Primary liver cancer is the sixth most common cancer worldwide, and it typically develops in the setting of cirrhosis. However, 20% of these cases can occur in non-cirrhotic livers [9][10].
Chronic hepatitis B and C, alcoholic liver disease and nonalcoholic steatohepatitis are the most common causes of liver cancer. Still, in some instances, HCC is detected incidentally during routine image examination with no definite aetiology.
Since altered BAs levels and their composition have a crucial role in HBCs development, it becomes obvious to think that sporadic primary liver cancers could be the result of mutations in genes involved in BAs transport and metabolism, especially in patients with no clear liver disease or with cryptogenic cholestasis.
The available data on inherited cholestatic diseases and HBCs, the old and the recently discovered cholestasis-related genes, the underlying pathophysiological mechanisms and finally, the interaction between gene mutations and environmental factors, as well as the enterohepatic circulation of BAs and the gut microbiota (Figure 1 summarizes genes, year of discovery and main phenotypes associated with the cholestatic diseases and the HBCs).
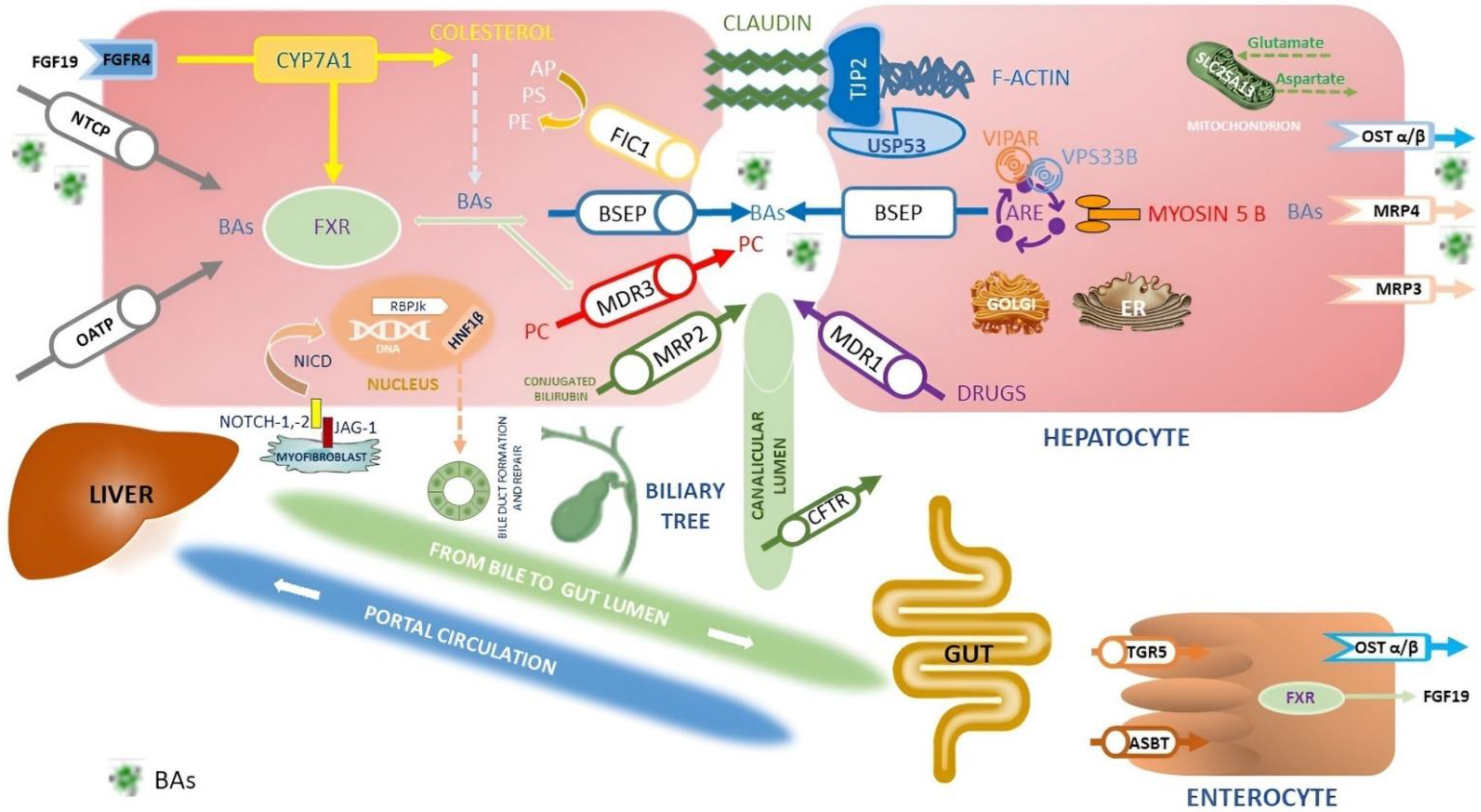
Figure 1. Simplified representation of the proteins expressed by the genes involved in cholestasis disorders and the main pathway of synthesis, transport and reuptake of BA in the liver and in the gut. Here researchers summarized the primary inherited cholestatic disorders in which metabolic and hepatobiliary diseases cause impaired BAs excretion. BAs are synthesized from cholesterol by CYP7A1 and then transported into canaliculi through the BSEP (PFIC2). Liver storage of BAs leads to liver injury, itching and increased risk of HBCs. Other constituents of bile include PC, transported by canalicular MDR3 (PFIC3), and PS, shuttled by canalicular ATP8B1 (PFIC1). Between disorders of membrane transporter or polarity, there is Dubin Johnson syndrome, where mutations in ABCC2 cause defects in MRP2, organic anions and bilirubin glucuronide transporter. Instead, ATP8B1 encodes MDR1 that translocates drugs and phospholipids across the hepatocyte membrane; it is responsible for developing resistance to anticancer drugs. The TJP2 (PFIC4), Claudine (neonatal ichthyosis sclerosing cholangitis), and USP53 (PFIC8) proteins are necessary to maintain the canalicular membrane polarity of hepatocytes and inhibit the reflux of BAs back into the cell: disorders of cytoskeletal and tight junction proteins cause cholestasis. The primary regulator of BAs metabolism is FXR (PFIC5): FXR inhibits CYP7A1 expression, stimulates the synthesis of FGF-19 to inhibit CYP7A1 expression through the FGFR4 pathway in the hepatocytes, stimulates BSEP to export of BAs, downregulates NTCP repressing the uptake of BAs by the liver, finally increases the expression of OST-α/β involved in BAs excretion from the liver to the portal vein and in intestinal reuptake. MYO5B, responsible for a form of PFIC and microvillous inclusion disease, interacts with RAB11A, altering the targeting of BSEP to the canalicular membrane via ARE; mutations in genes encoding RE-associated proteins such as MYO5B, VPS33B, and VIPAR (arthrogryposis, renal dysfunction and cholestasis syndrome—ARC) highlights the role of the RE in establishment and maintenance of hepatocyte polarity. BAs are carried into the hepatocyte by NTCP, OST-α, and OST-β on the basolateral membrane. Instead, ASBT on the ileal enterocyte reabsorbs approximately 95% of BAs, which enter the portal circulation via enterocyte transporters OST-α, OST-β, and MRP3. Mutations in SLC51A encoding the OSTα-OSTβ proteins cause PFIC6. Together with FXR, TGR5 is a primary BAS-sensing receptor involved in the interaction between BAs and microbiota; it is a negative regulator of the HCC envelope through its anti-inflammatory properties and abilities to guarantee correct homeostasis of BAs. SLC25A13 gene encodes the calcium-binding protein citrin, an aspartate–glutamate carrier sited within the inner mitochondrial membrane. Citrin plays a crucial role in protein, nucleotide, and urea synthesis in several metabolic pathways. Mutations in SLC25A13 lead to neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) and increased susceptibility to HBCs. Cell–cell Jagged/Notch interactions are critical for the differentiation of cells in the early phases of development. The ligand–receptor link induces proteolytic cleavage of the Notch receptor and release of the NICD. The NICD translocates into the nucleus where it activates RBPJκ, thus promoting Notch target genes’ transcription, including HNF1B. The persistent over-activation of Notch way in hepatic precursor cells leads to downstream RBPJk-dependent transcription activity, failing repair cell damage, induction of liver fibrosis and secondary HCC. Mutations in JAG1 and NOTCH2 genes are responsible for Alagille syndrome, while HNF1B is a target gene upregulated by NOTCH signalling. HNF1B regulates the differentiation of hepatoblasts into ductal plate cells and the inclusion of the developing duct into the portal space; mutations in the HNF1B gene have been associated with renal cysts and diabetes syndrome neonatal or late-onset cholestasis and some tumours, including liver cancer. CFTR (responsible for cystic fibrosis) is a chloride channel expressed by secretory epithelia, including the biliary epithelium in the liver. Furthermore, mutations affecting the function of CFTR can cause a cholestatic disorder: biliary architecture changes, severe sclerosing cholangitis, focal biliary cirrhosis and multi-lobular biliary cirrhosis complicated by portal hypertension are features of cystic fibrosis liver disease (CFLD). Finally, researchers omitted KIF12 in the figure since its localization in the Golgi apparatus and plasma membrane is uncertain: mutations in the KIF12 gene are associated with PFIC8. Abbreviations: ARE; apical recycling endosome; ASBT, apical sodium-dependent bile acid transporter; AP, amino-phospholipids; BAs, bile acids; BSEP, bile salt export pump protein; CFTR, cystic fibrosis transmembrane conductance regulator; CYP7A1, cholesterol 7α-monooxygenase; ER, endoplasmic reticulum; FGF19, fibroblast growth factor 19; FGFR14, fibroblast growth factor receptor 4; FIC 1, familial intrahepatic cholestasis deficiency type 1 protein; FXR, farnesoid X receptor; HNF-1B, Hepatocyte Nuclear Factor-1beta; JAG-2, Jagged Canonical Notch Ligand-2; KIF12, kinesin family member 12; MDR, multidrug resistance protein; MRP, multidrug resistance protein; NICD, Notch intracellular domain; NOTCH-1,2, Notch homolog-1,2 translocation-associated; NTCP, sodium taurocholate cotransporting polypeptide; OATP, organic anion transporting polypeptide; OST α/β, organic solute transporter alpha/beta; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PS, phosphatidylserine; PFIC, progressive familial intrahepatic cholestasis; RBPjk, recombining binding protein suppressor of hairless; SL25A13, solute carrier family 25 member 13; TGR5, G-protein-coupled bile acid receptor; TJP2, tight junction protein 2 gene; USP53, ubiquitin-specific peptidase 53; VIPAR, VPS33B interacting protein, apical–basal polarity regulator; VPS33B, vacuolar protein sorting associated protein 33B [6][7][11][12].
2. Progressive Familial Intrahepatic Cholestasis-Related Genes
2.1. Role of Next-Generation Sequencing
Traditional molecular testing methods greatly relied on Sanger sequencing technology [13]. So far, this technique is still the gold standard for molecular diagnosis when the genomic region of the disease is known and it is made of a few coding exons. It is also used for variants’ validation from high-throughput sequencing.
In the last years, NGS technologies have led to advances in understanding genome architecture [14]. NGS has dramatically facilitated the integration of NGS-based genetic analysis strategies in clinical diagnostics processes. In addition, it offers good data quality and affordable costs, improving data handling capabilities and increasing the computational power and efficiency of bioinformatics analysis tools. NGS can be used broadly for variants’ discovery. Panel gene sequencing (PGS) and WES/WGS are the main applications in molecular diagnosis.
PGS involves the selective enrichment of genes or genomic regions, already suggested by other genetic analyses and already known to be associated with clinical disorders or biological pathways. If disease genes are known and their number is limited (<100), it is recommended to set a target analysis with PGS. WES, instead, covers approximately the 20,000 known protein-coding genes in the human genome [13].
Mendelian and complex diseases are mainly caused (over 85%) by defects in protein sequences [15]. When classical target genes do not reveal any variation in their coding sequences, a WES approach is recommended since variations in coding sequences of other genes are probably present. Compared to other traditional molecular clinical diagnostic tests, WES presents higher rates of molecular diagnostic success when used for common disease traits, non-specific phenotypes, and rare variants [16][17][18].
WES technology is useful in the identification of rare and de novo mutations in the analysis of trios or families [19]. For instance, the other cholestasis-related genes have been identified by this approach.
The recent spread of “omics” studies, the complete sequencing of the human genome, and the decrease in the cost of this technology (less than USD 1000 per genome in the Illumina NovaSeq or BGI/MGI platforms) have favoured the use of WGS in research and clinical genetic diagnosis [14][20]. The T2T-CHM13 assembly added five complete chromosomal arms and more additional sequences than any genomic reference released in the last 20 years. While 8% of the human genome was omitted in the past due to technological limitations, now high-precision long-read sequencing has finally removed this technical barrier. This will make it possible in the future to rapidly discover new genes and new genetic pathways underlying disease and health [15].
Most Mendelian diseases are due to deleterious mutations within the exome. However, it is possible genetic variations having significant clinical implications can occur outside the exome sequences, risking not being identified [21][22]. Using WGS, it is also possible to find variants caused by copy number variants (CNVs), short tandem repeats (STRs), DNA deletions and insertions (INDELs) and structural variants (SVs).
2.2. Hepatobiliary Cancers and Cholestasis-Related Genes: An Underestimated Association
Hepatocytes are cells involved in many metabolic cell functions. They are, therefore, subject to many injuries potentially resulting in abnormal proliferation leading to liver cirrhosis as well as HCC.
Chronic hepatitis B and C are the most common etiologies associated with HCC in adults, followed by alcoholic and nonalcoholic steatohepatitis [23].
On the opposite to what is observed in adults, in children, HCC often develops in a non-cirrhotic setting. In Western countries, pediatric liver cancers were reported to occur in patients with PFIC; notably, 5–15% of children with PFIC2 present HCC early, from 13 to 28 months of age [23][24][25], but several cases are well described in infants with TJP2 disease (PFIC4) and MDR3 deficiency (PFIC3) [26][27][28][29].
In some instances, liver cancer is diagnosed incidentally during routine image exams, in young and adult people with unknown chronic liver disease, often because of mutations in critical genes involved in the metabolic pathways of hepatocytes or the metabolism and transport of BAs. As a result of ABCB4 variants or other mutations in cholestasis-related genes, these subjects often present serum liver enzyme abnormalities, particularly alanine aminotransferase, aspartate amino-transferase and GGT, in the absence of a clear aetiology of liver injury [1]. About 20% of cases of HCC can develop in a non-cirrhotic liver presenting with two peaks during the 2nd and 7th decade of life [9]. Unfortunately, in these patients, data, especially about genetic risk factors, are missing. A late-onset disease, a slower progression of a liver disorder or a predisposition to further hepatic injury are all possible phenotypic manifestations when genes involved in inherited cholestatic diseases are affected with a single variant (heterozygotic state) or homozygous variants with a milder reduction in protein function, or a combination of both [8]. Therefore, the attention is recently focusing on variants of these genes associated with childhood diseases among adults with different pictures of cryptogenic cholestasis [1].
Large-scale whole-genome sequencing of the Icelandic population showed a strong association between some mutations in the ABCB4 gene and the increased risk of liver diseases. In addition to gallstone disease, ICP, liver cirrhosis and increased serum liver-related exams (alanine transaminase, aspartate transaminase and GGT), the ABCB4 variants were also linked to liver, gallbladder and gall ways cancer. A total of 681 cases were diagnosed with HBCs in Iceland from 1955 to 2011, identified by the nationwide Icelandic Cancer Registry. Four variants in ABCB4 (c.1865G>A, p.G622E; c.1333_1334delCT, p.L445Gfs*22; c.1529A>G, p.N510S; c.711A>T, p.I237=) were associated with these liver-specific tumours [30].
Variants of ABCB4, together with those of the PNPLA3 gene for fatty liver disease, are considered the most relevant common genetic determinants of the cholestatic response of the liver to injury. Therefore, given the increased risk of developing HBCs, especially young subjects with known mutations in the ABCB4 gene, should undergo surveillance programs [31].
Interestingly, gene rearrangements in ABCB11 deficiency, using Mdr2-KO mice, frequently target the mitogen-activated protein kinase signalling pathway and stress the mechanisms leading from the gene defects to liver cancers secondary to phosphatidylcholine deficiency and low-phospholipid concentrations in bile. They play as inducers of liver injury in the general population and not only in patients with rare liver diseases such as PFIC, ICP or LPAC [32].
Moreover, polymorphisms such as rs2109505 (c.711A>T, p.I237=) in ABCB4 and rs2287622 (c.1331T>C, p.V444A) in ABCB11 are more prevalent in adult patients with idiopathic cholestasis than in healthy controls representing risk factors for the development of liver fibrosis [33]. In addition, the variant p.V444A in ABCB11 was observed more frequently in patients with DIC and ICP because of reduced liver BSEP expression [8].
In keeping with these findings, it could be speculated that many of the mild phenotypes concerning idiopathic cholestasis in adult people could be due to single or compound mutations in PFIC-related genes.
Pathogenic or likely pathogenic mutations in ATP8B1, ABCB11, ABCB4 and TJP2 genes can be responsible for 21% of idiopathic cholestasis in young and adult patients: they have higher rates of cholestatic histological features, higher levels of liver fibrosis and serum BAs compared to the subjects without, at least likely, pathogenic mutations [1].
The extensive development of diagnostic methods such as NGS and WES is leading to the discovery of numerous additional genes responsible for inherited cholestatic diseases and the application of historical gene panels related to child pathologies in the adult population with forms of cryptogenic liver disease.
It is unclear whether the prevalence of variants in cholestatic-related genes is as low in adults as in children.
Since some PFIC gene variants in ABCB11, ABCB4, and TJP2 are described in patients with liver tumours, subjects with idiopathic chronic cholestasis and personal or familial risk factors for inherited cholestasis as well as DIC, ICP or LPAC history, should be screened for a panel of primary cholestasis-related genes and might likely benefit from monitoring with periodic ultrasound exams.
References
- Vitale, G.; Gitto, S.; Raimondi, F.; Mattiaccio, A.; Mantovani, V.; Vukotic, R.; D’Errico, A.; Seri, M.; Russell, R.B.; Andreone, P. Cryptogenic cholestasis in young and adults: ATP8B1, ABCB11, ABCB4, and TJP2 gene variants analysis by high-throughput sequencing. J. Gastroenterol. 2018, 53, 945–958.
- Wu, L.; Feng, J.; Li, J.; Yu, Q.; Ji, J.; Wu, J.; Dai, W.; Guo, C. The gut microbiome-bile acid axis in hepatocarcinogenesis. Biomed. Pharm. 2020, 133, 111036.
- Yamada, S.; Takashina, Y.; Watanabe, M.; Nagamine, R.; Saito, Y.; Kamada, N.; Saito, H. Bile acid metabolism regulated by the gut microbiota promotes non-alcoholic steatohepatitis-associated hepatocellular carcinoma in mice. Oncotarget 2018, 9, 9925–9939.
- Jia, B.; Jeon, C.O. Promotion and induction of liver cancer by gut microbiome-mediated modulation of bile acids. PLoS Pathog. 2019, 15, e1007954.
- Ibrahim, S.H.; Kamath, B.M.; Loomes, K.M.; Karpen, S.J. Cholestatic liver diseases of genetic etiology: Advances and controversies. Hepatology 2022, 75, 1627–1646.
- Fabris, L.; Fiorotto, R.; Spirli, C.; Cadamuro, M.; Mariotti, V.; Perugorria, M.J.; Banales, J.M.; Strazzabosco, M. Pathobiology of inherited biliary diseases: A roadmap to understand acquired liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 497–511.
- Goldberg, A.; Mack, C.L. Inherited Cholestatic Diseases in the Era of Personalized Medicine. Clin. Liver Dis. 2020, 15, 105–109.
- Nayagam, J.S.; Williamson, C.; Joshi, D.; Thompson, R.J. Review Article: Liver Disease in Adults with Variants in the Cho-lestasis-Related Genes ABCB11, ABCB4 and ATP8B1. Aliment. Pharmacol. Ther. 2020, 52, 1628–1639.
- Desai, A.; Sandhu, S.; Lai, J.-P.; Sandhu, D.S. Hepatocellular carcinoma in non-cirrhotic liver: A comprehensive review. World J. Hepatol. 2019, 11, 1–18.
- Lee, D.H.; Lee, J.M. Primary malignant tumours in the non-cirrhotic liver. Eur. J. Radiol. 2017, 95, 349–361.
- Kriegermeier, A.; Green, R. Pediatric Cholestatic Liver Disease: Review of Bile Acid Metabolism and Discussion of Current and Emerging Therapies. Front. Med. 2020, 7, 149.
- Li, Q.; Sun, Y.; van Ijzendoorn, S.C.D. A Link between Intrahepatic Cholestasis and Genetic Variations in Intracellular Trafficking Regulators. Biology 2021, 10, 119.
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467.
- Schuster, S.C. Next-generation sequencing transforms today’s biology. Nat. Methods 2007, 5, 16–18.
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53.
- Kryukov, G.V.; Pennacchio, L.A.; Sunyaev, S.R. Most Rare Missense Alleles Are Deleterious in Humans: Implications for Complex Disease and Association Studies. Am. J. Hum. Genet. 2007, 80, 727–739.
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical Whole-Exome Sequencing for the Diagnosis of Mendelian Disorders. N. Engl. J. Med. 2013, 369, 1502–1511.
- Drew, A.P.; Zhu, D.; Kidambi, A.; Ly, C.; Tey, S.; Brewer, M.H.; Ahmad-Annuar, A.; Nicholson, G.A.; Kennerson, M.L. Improved inherited peripheral neuropathy genetic diagnosis by whole-exome sequencing. Mol. Genet. Genom. Med. 2015, 3, 143–154.
- Long, P.A.; Evans, J.M.; Olson, T.M. Exome sequencing establishes diagnosis of Alström syndrome in an infant presenting with non-syndromic dilated cardiomyopathy. Am. J. Med Genet. Part A 2015, 167, 886–890.
- Zhang, X. Exome sequencing greatly expedites the progressive research of Mendelian diseases. Front. Med. 2014, 8, 42–57.
- Rexach, J.; Lee, H.; Martinez-Agosto, J.A.; Németh, A.H.; Fogel, B.L. Clinical application of next-generation sequencing to the practice of neurology. Lancet Neurol. 2019, 18, 492–503.
- Guo, Y.; Long, J.; He, J.; Li, C.-I.; Cai, Q.; Shu, X.-O.; Zheng, W.; Li, C. Exome sequencing generates high quality data in non-target regions. BMC Genom. 2012, 13, 194.
- Brar, T.S.; Hilgenfeldt, E.; Soldevila-Pico, C. Etiology and Pathogenesis of Hepatocellular Carcinoma. In Precision Molecular Pathology of Liver Cancer; Liu, C., Ed.; Molecular Pathology Library; Springer International Publishing: Cham, Switzerland, 2018; pp. 1–15. ISBN 978-3-319-68082-8.
- Knisely, A.S.; Strautnieks, S.S.; Meier, Y.; Stieger, B.; Byrne, J.A.; Portmann, B.C.; Bull, L.N.; Pawlikowska, L.; Bilezikçi, B.; Özçay, F.; et al. Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology 2006, 44, 478–486.
- Davit-Spraul, A.; Fabre, M.; Branchereau, S.; Baussan, C.; Gonzales, E.; Stieger, B.; Bernard, O.; Jacquemin, E. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): Phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology 2010, 51, 1645–1655.
- Pawlikowska, L.; Strautnieks, S.; Jankowska, I.; Czubkowski, P.; Emerick, K.; Antoniou, A.; Wanty, C.; Fischler, B.; Jacquemin, E.; Wali, S.; et al. Differences in presentation and progression between severe FIC1 and BSEP deficiencies. J. Hepatol. 2010, 53, 170–178.
- Zhou, S.; Hertel, P.M.; Finegold, M.J.; Wang, L.; Kerkar, N.; Wang, J.; Wong, L.C.; Plon, S.E.; Sambrotta, M.; Foskett, P.; et al. Hepatocellular carcinoma associated with tight-junction protein 2 deficiency. Hepatology 2015, 62, 1914–1916.
- Sambrotta, M.; Strautnieks, S.; Papouli, E.; Rushton, P.; Clark, B.E.; Parry, D.A.; Logan, C.V.; Newbury, L.J.; Ka-math, B.M.; Ling, S.; et al. Mutations in TJP2 cause progressive cholestatic liver disease. Nat. Genet. 2014, 46, 326–328.
- Vij, M.; Shanmugam, N.P.; Reddy, M.S.; Govil, S.; Rela, M. Hepatocarcinogenesis in multidrug-resistant P-glycoprotein 3 deficiency. Pediatr. Transpl. 2017, 21, e12889.
- Gudbjartsson, D.F.; Helgason, H.; Gudjonsson, S.A.; Zink, F.; Oddsson, A.; Gylfason, A.; Besenbacher, S.; Magnusson, G.; Halldórsson, B.; Hjartarson, E.; et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 2015, 47, 435–444.
- Lammert, F.; Hochrath, K. A letter on ABCB4 from Iceland: On the highway to liver disease. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 655–658.
- Iannelli, F.; Collino, A.; Sinha, S.K.; Radaelli, E.; Nicoli, P.; D’Antiga, L.; Sonzogni, A.; Faivre, J.; Buendia, M.A.; Sturm, E.; et al. Massive gene amplification drives paediatric hepatocellular carcinoma caused by bile salt export pump deficiency. Nat. Commun. 2014, 5, 3850.
- Jüngst, C.; Justinger, C.; Fischer, J.; Berg, T.; Lammert, F. Common ABCB4 and ABCB11 Genotypes Are Associated with Idiopathic Chronic Cholestasis in Adults. Dig. Dis. 2021, 40, 489–496.
More
Information
Subjects:
Gastroenterology & Hepatology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
970
Revisions:
2 times
(View History)
Update Date:
24 Feb 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No