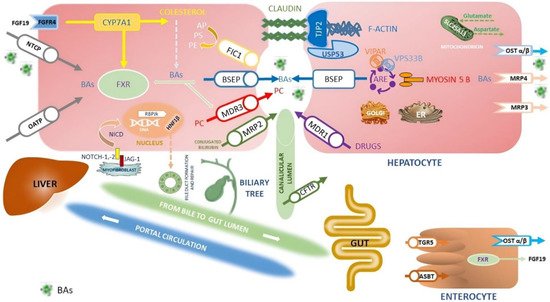
Figure 1. Simplified representation of the proteins expressed by the genes involved in cholestasis disorders and the main pathway of synthesis, transport and reuptake of BA in the liver and in the gut. Here
rwe
searchers summarized the primary inherited cholestatic disorders in which metabolic and hepatobiliary diseases cause impaired BAs excretion. BAs are synthesized from cholesterol by CYP7A1 and then transported into canaliculi through the BSEP (PFIC2). Liver storage of BAs leads to liver injury, itching and increased risk of HBCs. Other constituents of bile include PC, transported by canalicular MDR3 (PFIC3), and PS, shuttled by canalicular ATP8B1 (PFIC1). Between disorders of membrane transporter or polarity, there is Dubin Johnson syndrome, where mutations in
ABCC2 cause defects in MRP2, organic anions and bilirubin glucuronide transporter. Instead,
ATP8B1 encodes MDR1 that translocates drugs and phospholipids across the hepatocyte membrane; it is responsible for developing resistance to anticancer drugs. The TJP2 (PFIC4), Claudine (neonatal ichthyosis sclerosing cholangitis), and USP53 (PFIC8) proteins are necessary to maintain the canalicular membrane polarity of hepatocytes and inhibit the reflux of BAs back into the cell: disorders of cytoskeletal and tight junction proteins cause cholestasis. The primary regulator of BAs metabolism is FXR (PFIC5): FXR inhibits CYP7A1 expression, stimulates the synthesis of FGF-19 to inhibit CYP7A1 expression through the FGFR4 pathway in the hepatocytes, stimulates BSEP to export of BAs, downregulates NTCP repressing the uptake of BAs by the liver, finally increases the expression of OST-α/β involved in BAs excretion from the liver to the portal vein and in intestinal reuptake. MYO5B, responsible for a form of PFIC and microvillous inclusion disease, interacts with RAB11A, altering the targeting of BSEP to the canalicular membrane via ARE; mutations in genes encoding RE-associated proteins such as MYO5B, VPS33B, and VIPAR (arthrogryposis, renal dysfunction and cholestasis syndrome—ARC) highlights the role of the RE in establishment and maintenance of hepatocyte polarity. BAs are carried into the hepatocyte by NTCP, OST-α, and OST-β on the basolateral membrane. Instead, ASBT on the ileal enterocyte reabsorbs approximately 95% of BAs, which enter the portal circulation via enterocyte transporters OST-α, OST-β, and MRP3. Mutations in
SLC51A encoding the OSTα-OSTβ proteins cause PFIC6. Together with FXR, TGR5 is a primary BAS-sensing receptor involved in the interaction between BAs and microbiota; it is a negative regulator of the HCC envelope through its anti-inflammatory properties and abilities to guarantee correct homeostasis of BAs.
SLC25A13 gene encodes the calcium-binding protein citrin, an aspartate–glutamate carrier sited within the inner mitochondrial membrane. Citrin plays a crucial role in protein, nucleotide, and urea synthesis in several metabolic pathways. Mutations in
SLC25A13 lead to neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) and increased susceptibility to HBCs. Cell–cell Jagged/Notch interactions are critical for the differentiation of cells in the early phases of development. The ligand–receptor link induces proteolytic cleavage of the Notch receptor and release of the NICD. The NICD translocates into the nucleus where it activates RBPJκ, thus promoting Notch target genes’ transcription, including
HNF1B. The persistent over-activation of Notch way in hepatic precursor cells leads to downstream RBPJk-dependent transcription activity, failing repair cell damage, induction of liver fibrosis and secondary HCC. Mutations in
JAG1 and
NOTCH2 genes are responsible for Alagille syndrome, while
HNF1B is a target gene upregulated by NOTCH signalling.
HNF1B regulates the differentiation of hepatoblasts into ductal plate cells and the inclusion of the developing duct into the portal space; mutations in the
HNF1B gene have been associated with renal cysts and diabetes syndrome neonatal or late-onset cholestasis and some tumours, including liver cancer. CFTR (responsible for cystic fibrosis) is a chloride channel expressed by secretory epithelia, including the biliary epithelium in the liver. Furthermore, mutations affecting the function of CFTR can cause a cholestatic disorder: biliary architecture changes, severe sclerosing cholangitis, focal biliary cirrhosis and multi-lobular biliary cirrhosis complicated by portal hypertension are features of cystic fibrosis liver disease (CFLD). Finally,
rwe
searchers omitted KIF12 in the figure since its localization in the Golgi apparatus and plasma membrane is uncertain: mutations in the
KIF12 gene are associated with PFIC8. Abbreviations: ARE; apical recycling endosome; ASBT, apical sodium-dependent bile acid transporter; AP, amino-phospholipids; BAs, bile acids; BSEP, bile salt export pump protein; CFTR, cystic fibrosis transmembrane conductance regulator; CYP7A1, cholesterol 7α-monooxygenase; ER, endoplasmic reticulum; FGF19, fibroblast growth factor 19; FGFR14, fibroblast growth factor receptor 4; FIC 1, familial intrahepatic cholestasis deficiency type 1 protein; FXR, farnesoid X receptor; HNF-1B, Hepatocyte Nuclear Factor-1beta; JAG-2, Jagged Canonical Notch Ligand-2; KIF12, kinesin family member 12; MDR, multidrug resistance protein; MRP, multidrug resistance protein; NICD, Notch intracellular domain; NOTCH-1,2, Notch homolog-1,2 translocation-associated; NTCP, sodium taurocholate cotransporting polypeptide; OATP, organic anion transporting polypeptide; OST α/β, organic solute transporter alpha/beta; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PS, phosphatidylserine; PFIC, progressive familial intrahepatic cholestasis; RBPjk, recombining binding protein suppressor of hairless; SL25A13, solute carrier family 25 member 13; TGR5, G-protein-coupled bile acid receptor; TJP2, tight junction protein 2 gene; USP53, ubiquitin-specific peptidase 53; VIPAR, VPS33B interacting protein, apical–basal polarity regulator; VPS33B, vacuolar protein sorting associated protein 33B
[6][7][11][12][6,7,12,13].