Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jm Campagne | -- | 3352 | 2023-02-21 14:13:11 | | | |
| 2 | Jason Zhu | Meta information modification | 3352 | 2023-02-22 03:26:07 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Taussat, A.; De Figueiredo, R.M.; Campagne, J. Direct Amidations of Carboxylic Acids with Amines. Encyclopedia. Available online: https://encyclopedia.pub/entry/41482 (accessed on 22 July 2026).
Taussat A, De Figueiredo RM, Campagne J. Direct Amidations of Carboxylic Acids with Amines. Encyclopedia. Available at: https://encyclopedia.pub/entry/41482. Accessed July 22, 2026.
Taussat, Armand, Renata Marcia De Figueiredo, Jean-Marc Campagne. "Direct Amidations of Carboxylic Acids with Amines" Encyclopedia, https://encyclopedia.pub/entry/41482 (accessed July 22, 2026).
Taussat, A., De Figueiredo, R.M., & Campagne, J. (2023, February 21). Direct Amidations of Carboxylic Acids with Amines. In Encyclopedia. https://encyclopedia.pub/entry/41482
Taussat, Armand, et al. "Direct Amidations of Carboxylic Acids with Amines." Encyclopedia. Web. 21 February, 2023.
Copy Citation
The prevalence of amides in biological systems and chemical fields such as polymers, materials and natural products drives continuous research on novel procedures to obtain these ubiquitous functional groups. Efforts to this purpose are mainly focused around the discovery of direct and catalytic methods that are more atom economic, safe and practical for diversified applications (e.g., organic, medicinal and peptide chemistries, material and polymer purposes, etc.), in accordance with green chemistry principles.
amides
metal-transition catalysis
direct aminolysis
1. Introduction
The amide bond is of particular importance, not only for its key function in peptide/protein structures, but also its role in a large number of natural and synthetic small molecules and polymers [1][2]. Amide bond formation is traditionally achieved through the activation of the carboxylic acid partner using a greater-than-stoichiometric quantity of some complex activating agent (carbodiimides + additives (HOBt, HOAt or Oxyma), phosphonium or guanidinium salts, etc.), thus generating a large amount of unvalorized byproducts, whereas amide formation is ‘just’ about the elimination of one molecule of water. In 2006, a round table dedicated to the development of green chemistry research ranked “amide formation avoiding poor atom economy reagents” as a top priority [3]. This point is even more crucial, as the formation of amides from acids and amines is by far the most used reaction in medicinal chemistry [4]. This intends to highlight more recent progress in catalytic formation of amide bonds from amines, carboxylic acids and esters (Scheme 1) [5][6][7]. These catalytic amidation strategies were recently covered by Sheppard, Wang and Perrin, who focused on green aspects, perspectives and peptide synthesis [8][9][10]. General reviews on amide bond formation that do not focus on catalytic direct approaches have also appeared [11][12].
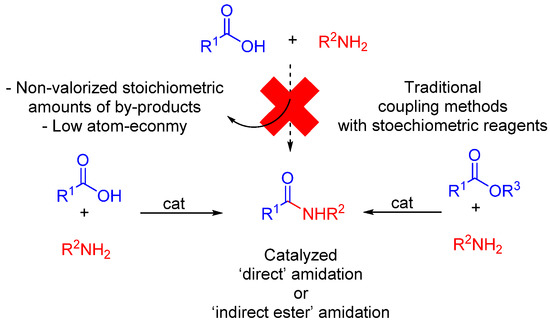
Scheme 1. Catalytic amidation reactions from amines and acids (or esters).
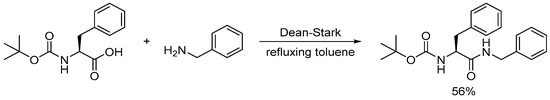
Before discussing catalyzed reactions, it is appropriate to comment on amidation reactions under thermal conditions in the absence of a catalyst. The direct thermal condensation of acids and amines is indeed possible, but usually requires high temperatures (>160 °C) to proceed and is thus generally limited to insensitive and poorly functionalized substrates [13][14]. The key issue is the formation of poorly reactive ammonium salts from the carboxylic and amine partners. Nevertheless, these statements must be tempered and depend largely on the reaction partners, in particular, the acidity of the carboxylic acid, and the reaction conditions [15]. Therefore, when a catalytic reaction is carried out under thermal conditions, it is necessary to conduct a control reaction without any catalyst to obtain the background uncatalyzed reaction. For example, the reaction of Boc-Phe-OH with benzylamine under refluxing toluene using a Dean–Stark apparatus allows the formation of the corresponding amide at a 56% yield (Scheme 2) [16]. Microwave-assisted solvent-free preparations have appeared in the literature and may be considered as practical and easy to handle conditions for undemanding substrates [17][18].
Scheme 2. Thermal amidation of Boc-Phe-OH with benzylamine.
In direct amidation reactions from amines and carboxylic acids, the only byproduct is water. Thus, the central question is how to trap/remove water to promote/favor amide formation. Either azeotropic removal of water (using a Dean–Stark apparatus or a Soxhlet extractor with a dehydrating agent) or excess molecular sieves have been used. While molecular sieves allow a reaction at lower temperature, they are incompatible with large-scale synthesis. Indeed, catalytic amidations have been used only marginally in the large-scale synthesis of amides [19].
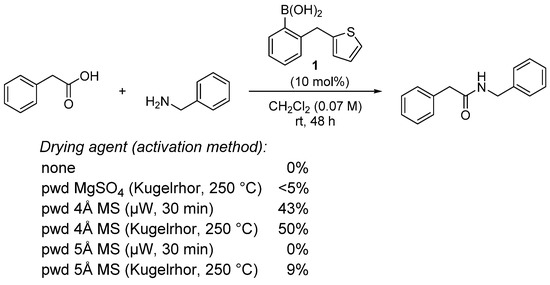
The nature of the dehydrating agent and the method of activating it may also be crucial in these reactions, as illustrated by Blanchet in the boronic acid 1-catalyzed amidation of phenylacetic acid with benzylamine (Scheme 3) Supplementary Information described in ref [20]. Both the type of drying agent (4 Å MS vs. 5 Å MS) and the activation method (microwaves vs. Kugelrhor) have a profound impact on the yield. Questions as to the actual role of molecular sieves in these reactions have thus arisen, and recent developments have shed light on the function of these agents beyond simple water traps (Scheme 3).
Scheme 3. Influence of the nature and method of activation of the drying agent in an amidation reaction; µM = microwaves.
2. Boron-Derived Catalysts
From the seminal boronic acid catalysts described by Yamamoto in 1996 (structure 2) to the much more elaborated bis-boronic acids (Figure 1, structures 3–11), boron derivatives have emerged as an efficient class of catalysts that allow the formation of amides under milder conditions in the presence of sensitive and functionalized substrates. For a general review of boronic acid catalysis, see [21].
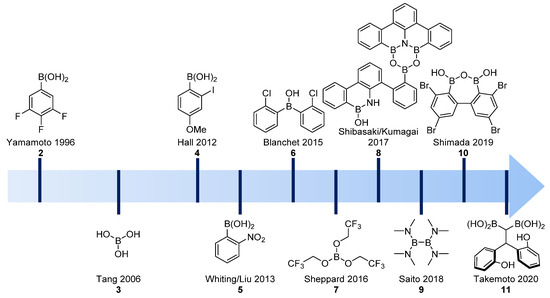
Figure 1. Evolution of boron-derived catalysts used in direct amidation reactions.
Since the publication of Ishihara and Yamamoto’s seminal paper, more than 20 boron-derived catalysts were described in the literature in the 1996–2015 period (for a list of catalysts developed in this period, see [5]). Initially, in 1996, refluxing conditions were required (using toluene, xylene or mesitylene), and thus these reactions were restricted to rather non-functionalized substrates.
As illustrated in Figure 1, ortho-substituted derivatives (see structures 4–6, Figure 1) have experimentally emerged as promising catalytic systems, allowing reactions at room temperature for less-demanding substrates and reactions around 60 °C for the synthesis of dipeptides [20][22][23]. Ishihara also described the use of DMAPO as an efficient acyl-transfer co-catalyst in these reactions, as illustrated in the multigram-scale of N-Boc-protected sitaglipin [24][25] (Scheme 4). This co-catalyst was also used in the microwave-assisted synthesis of cinnamides using a PhB(OH)2/DMAPO catalytic system [26].
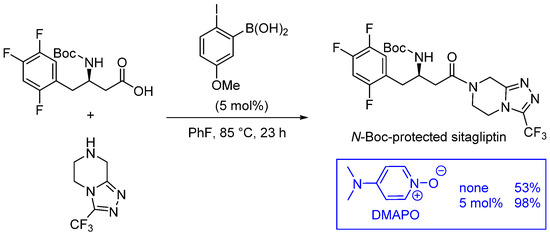
Scheme 4. DMAPO as a co-catalyst in boronic acid-catalyzed amidation reactions.
These ortho-substituted boronic acids have thus appeared experimentally as efficient catalysts, but their efficiency has not been directly correlated to the generally accepted mechanism for these reactions. A major breakthrough in this area was achieved in 2018 in an extensive mechanistic study by Sheppard and Whiting [27]. Supported by related analytical, experimental and theoretical studies, evidence has been found that the reaction does not proceed through a ‘monocyclic’ acyloxyboron 12 active intermediate, as was initially proposed and accepted, but through the formation of a bicyclic intermediate involving a 2:2 carboxylic acid/arylboronic complex 13 (or 14) (Scheme 5b). Such intermediates embed a B-X-B (X = O or NR) connection, as previously observed by X-ray analysis in tetraacetyl diborate, which was originally assigned a triacetoxyborate structure (Scheme 5a).
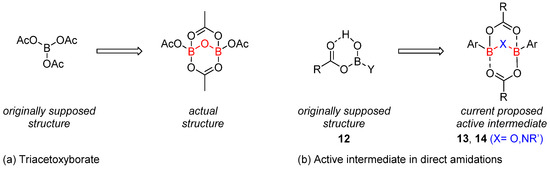
Scheme 5. Mono- vs. bicyclic structures of boron derivatives. (a) Triacetoxyborate, (b) Active intermediate in direct amidations.
The isolated (and fully characterized) bicyclic derivative 13 is a key active intermediate in amide formation. In the presence of an amine, a new intermediate can be observed in 11B NMR, which was tentatively attributed to the bicyclic compound 14 (Scheme 6). From the intermediates 13 or 14, different mechanisms to explain the formation of the amide bond have been studied computationally, but no preferential pathway has been identified.
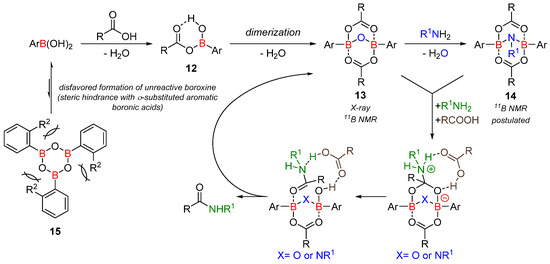
Scheme 6. Postulated (simplified) amidation mechanism.
The study by Sheppard and Whiting may also shed lights on some yet unexplained experimental observations:
-
The formation of the bicyclic intermediate 13 is highly dependent of the quantity and nature of molecular sieves; a large quantity of 5 Å MS is required and less than 5% conversion was observed with 4 Å MS (see Scheme 3);
-
Boroxine 15 could be considered as a poorly reactive ‘resting state’ intermediate. As determined by DFT, the use of o-substituted aromatic boronic acids destabilizes the formation of the corresponding boroxine 15 derivatives, thus explaining the efficiency and success of such ortho-substituted catalysts (see Figure 1);
-
Finally, it should be noted that for borinic acid 6, the actual catalyst can be determined to be ortho-chloro boronic acid (obtained by protodeborylation of borinic acid 6).
Moreover, this mechanistic study is in line with a work published in 2017 by Shibasaki and Kumagai describing the use of B-O-B heterocycles as efficient catalysts for direct amidation reactions [28], where 1,3-Dioxa-5-aza-2,4,6-triborinane (DATB) derivative 8 was found to be an efficient catalyst for the amidation of a wide range of carboxylic acids and amines. The scope of this catalyst includes undemanding substrates (for which catalytic loadings of 0.5 mol% can be used), highly epimerizable ones and hindered substrates or APIs, and the yields are high (Scheme 7).
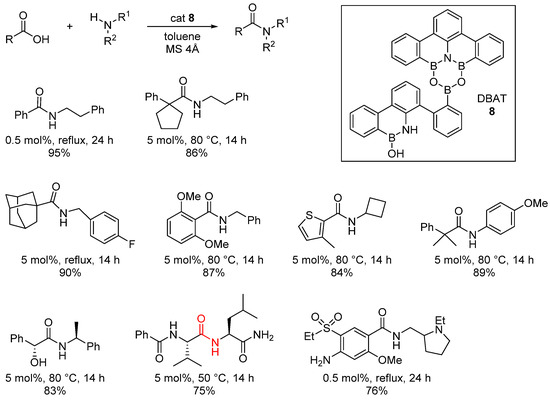
Scheme 7. DTAB 8 catalyzed reactions.
This methodology has also been applied to the synthesis of peptides using Fmoc amino acids. This synthetic potential was illustrated in the synthesis of a pentapeptide (obtained by fragment coupling at the C-terminus of a glycine residue) [29].
The major drawback of this methodology is the lengthy route to obtain such DTAB derivatives. Kumagai and Shibasaki then developed pyrimidine analogs—so-called Pym-DTABs (Figure 2). These compounds are more accessible and show rather similar catalytic performance (temperature, loading, scope) [30].
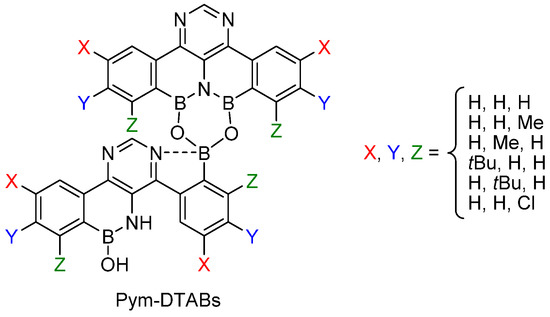
Figure 2. Pym-DTAB structure.
Mechanistic studies have highlighted the dual role of the two distinct classes of boron atoms [31]. The most Lewis acidic “O-B-O” boron atoms readily activate the amine via an adduct formation. The “B-N-B” moiety reacts as both a Brønsted base and a Lewis acid, allowing the formation of a “B-O=C-O-B” cyclic complex where the electrophilicity of the carboxylic function is activated (Scheme 8).
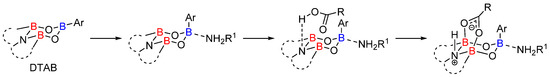
Scheme 8. Intermediates involved in the DTAB-catalyzed reactions.
Saito also described the use of simpler diboron structures—tetrahydroxy diboron and tetrakis(dimethylamino) diboron 9—to enable the catalytic amidation of various (hetero)aromatic acids in refluxing toluene, with good yields with low catalytic loadings (2 mol%) [32]. As illustrated in Figure 3, the cooperative role of the two boron centers in the amidation process is highlighted by structure 16, detected by ESI-HRMS.
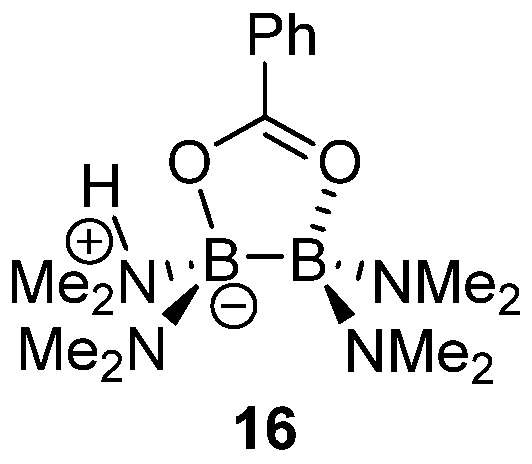
Figure 3. Mechanistic insights into the carboxylic acid activation with diboron catalyst 9.
Shimada next developed diboronic acid anhydride 10 (Figure 1) with a B-O-B linkage. The catalyst 10 was shown to be effective in the amidation of alpha or beta hydroxy acids with a wide range of amines or anilines, but not in cases of ‘simple’ carboxylic acids (without OH groups). It also should be noted that these reactions were carried out in toluene (60 °C or reflux) in the absence a molecular sieve or Dean–Stark apparatus [33]. The reaction was next extended to β-hydroxy-α-amino acids (serine, threonine derivatives, etc.), to the formation of Weinreb amides and the synthesis of Garner’s aldehyde [34][35][36].
In 2020, Takemoto analyzed the catalytic performance of gem-diboronic acid 11, incorporating a B-C-B linkage, in catalytic dehydrative peptide synthesis. As illustrated in Scheme 9, dipeptides were obtained in good yields in toluene at 65 °C in the presence of 5 Å MS. Epimerization-prone dipeptides were also synthesized, with only partial epimerization observed [37].
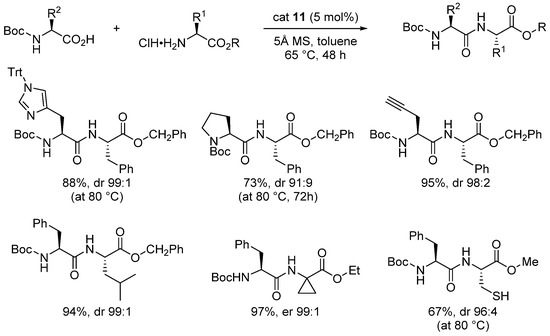
Scheme 9. Scope of dipeptide synthesis using cat 11.
Preliminary mechanistic experiments were carried out. Cat 11 is not the actual catalyst and rapidly evolved in the presence of 5 Å MS to form of cis-17 (determined by X-ray analysis). Addition of Boc-Phe-OH (in the presence of a proton sponge) led to the observation (by ESI-MS) of the dehydrated complex 18, which embeds the two boron atoms and the carboxylic acid (Scheme 10).
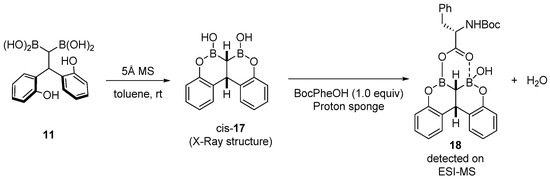
Scheme 10. Mechanistic insights into cat 11-catalyzed reactions.
The fine comprehension of the reaction mechanisms and the search for gentle reaction conditions allowing the coupling of sensitive and/or functionalized substrates has led to the development of more and more structurally complex catalysts. More simple catalysts have also emerged, such as borate B(OCH2CF3)3 7 developed by Sheppard [38][39]. In particular, Sheppard achieved an impressive selective catalytic amidation of unprotected amino acids. A large range of unprotected amino-acids and amines can be used in these reactions, with tert-amyl methyl ether (TAME) as solvent at 86 °C and a Dean–Stark apparatus [40]. As illustrated by the authors, the key point is the formation of soluble intermediate 19 in minute amounts from the insoluble zwitterionic amino acid (Scheme 11). This intermediate, in the presence of an apparent excess of amine, will smoothly yield the desired amide, while unwanted oligomerization is prevented. It should be emphasized that Ti(OiPr)4 could also be considered (at least in the most favorable cases) as a cheaper alternative in these reactions.
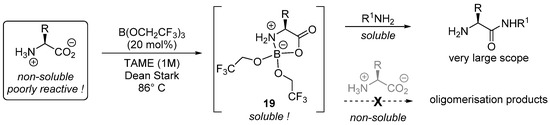
Scheme 11. Mechanistic insights into cat 7-catalyzed reactions; TAME = tert-amyl methyl ether.
Sheppard and co-workers also developed a one-pot sequential double amidation process. As illustrated in Scheme 12, good yields were usually observed, but with significant enantiopurity erosion.
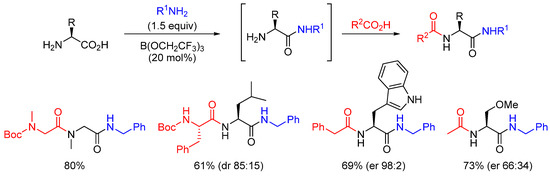
Scheme 12. Double amidation reactions.
More recently, the use of tert-butyl acetate as the solvent reaction was described by Sheppard. Beyond increased safety and durability, greater range was obtained using fewer nucleophilic anilines and polar substrates [41]. Interestingly, a large-scale procedure (100 mol) was described with no aqueous work-up or chromatography.
Nevertheless, it should be remembered that for undemanding substrates with low sensitivity, reactions can be carried out with simple and inexpensive boric acid [42][43][44], as recently illustrated by Popowycz for the synthesis of various amides from renewable isosorbide (Scheme 13) [45].
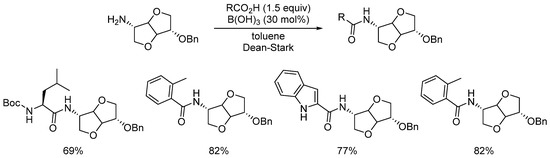
Scheme 13. Boric acid-catalyzed amidation of isosorbide.
3. Phosporus- and Silicon-Derived Catalysts
Since the seminal work of Chan [46][47], the use of silicon reagents to perform amidation reactions has been well known, but even in modern developments, these reactions still require a stoichiometric amount of a silicon reagent, resulting in poor atom economy [48][49]. In 2022, Haas described a solvent-free procedure using 20 mol% of dodecamethoxyneopentasilane 20 as catalyst [50]. As illustrated in Scheme 14, relatively poorly functionalized amides were obtained in good to excellent yields (13 examples) in the absence of any external water trap. Neopentasilane 20 (20 mol%) fragments into five reactive silicons upon methanolysis. In this reaction, water is trapped by the formation of a polysiloxane byproduct.
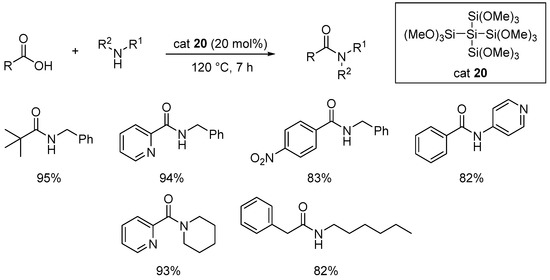
Scheme 14. Dodecamethoxyneopentasilane-catalyzed amidation reactions.
It should also be mentioned that the direct amidation of carboxylic acids can be performed under microwave irradiation using silica gel as a solid support and catalyst [51]. In a parallel study, the mechanism of this transformation was elucidated using IR and computational studies.
Phosphine and phospine oxide (organo)catalysts have also been described as promoting direct amidation reactions. They are mainly based on a P(III) ↔
P(V) catalytic cycle, which requires a reductant (or an oxidant) in stoichiometric quantity to regenerate the catalytic species [52][53][54].
In 2020, Sato described the use of tris(o-phenylenedioxy)cyclotriphosphazene TAP as a pre-catalyst for the direct amidation of aromatic acids and amines [55]. On the basis of 31P NMR and ESI-MS experiments, different relevant catalytic intermediates were identified (Scheme 15). TAP acts as a pre-catalyst, generating the catechol cyclic phosphate CCP in situ. This catalytic species promotes amide formation while generating pyrocatechol phosphate (PP) as a byproduct. Next, dehydration of PP regenerates CCP, thus completing the catalytic cycle. Regarding the formation of the amide bond, two mechanisms were considered by the authors, involving either an inner P-sphere mechanism (direct attack of the carboxylic partner on the phosphorus atom of the CCP) or an outer P-sphere (with the phosphate of the CCP acting to stabilize a tetrahedral intermediate) mechanism. It should be stressed that CCP is a poorly stable compound, which is why pre-catalysts such as TAP are required and reactions must be carried out under an inert atmosphere.
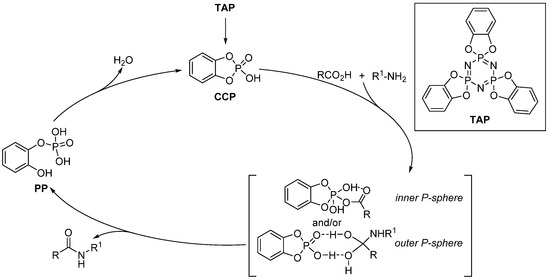
Scheme 15. Mechanistic rationale in TAP-catalyzed amidation reactions.
As illustrated in Scheme 16, a wide range (25 examples) of aromatic and heteroaromatic carboxylic acids have been efficiently coupled with mono- and di-substituted amines (Scheme 16). However, low yields were observed when aniline or H-Trp-OMe were used. Moreover, in the latter case, substantial erosion of the enantioselectivity (91:9 er) was also observed, probably due to the high temperature required in these reactions.
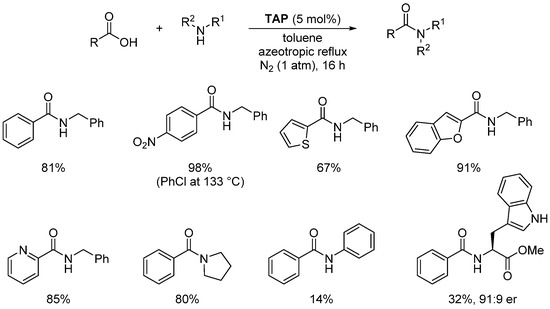
Scheme 16. TAP-catalyzed amidation reactions.
4. Organo-Catalyzed Reactions
Stoichiometric amounts of phosphines (or phosphites) have also been used in conceptually different organocatalyzed redox amidation reactions, playing on the cleavage of weak heteroatom–heteroatom bonds (S-N or Se-Se) of compounds 21–24 (Figure 4) [56][57][58][59].
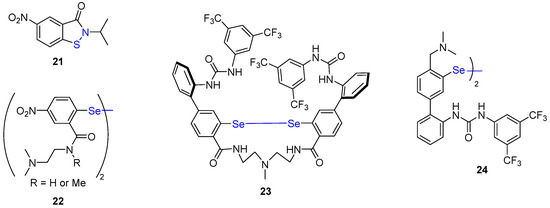
Figure 4. S-N- and Se-Se-based organocatalysts.
A general (and simplified) modus operandi is presented in Scheme 17. Cleavage of the weak heteroatom–heteroatom (S-N or Se-Se) bond by the nucleophilic phosphine enables the formation of an electrophilic phosphonium salt that can be trapped by the carboxylic acid. Such a generated activated acid can then react with the amine to generate the amide bond and phosphine oxide as a byproduct. Ultimately, the reduced organocatalyst is re-oxidized in situ to regenerate the catalyst and complete the catalytic cycle.
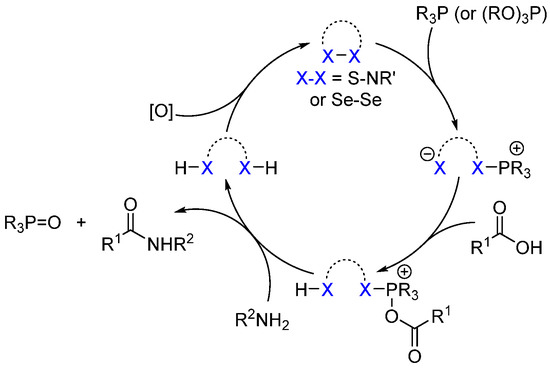
Scheme 17. General mechanism using S-N- and Se-Se-based organocatalysts.
S-acylthiosalicylamide 21 was first described by Liebeskind and notably allows the formation of various peptides and peptide derivatives under relatively mild conditions (20 mol% 21, (EtO)3P (1.5 equiv.), MeCN, 50 °C, dry air, 4 Å MS) with high yields and in the absence of racemization [56]. In this case, dry air was used in the presence of a catalytic amount of a copper to promote the regeneration of the organocatalyst 21.
Liebeskind extended this dehydrative strategy to the use of diselenide derivative 22 (R = Me or H) [57]. The use of selenium instead of sulfur was chosen to enhance the re-oxidation of the reduced form of the organocatalyst and thus avoid the use of copper as a co-catalyst. Using diselenide 22, catalyst loading can be reduced to 2.5 mol% and the reaction can be carried out in the absence of copper.
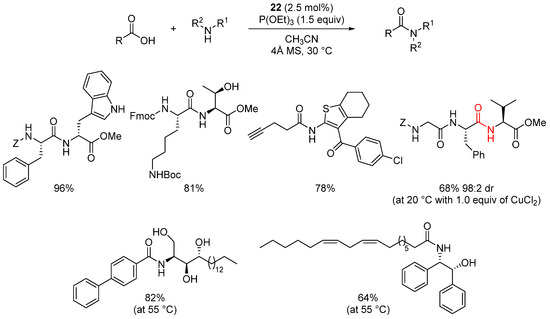
Scheme 18. Organocatalyzed 22 amidation reactions.
The design of the diselenide organocatalyst was next refined by Arora, introducing a urea in 23 to stabilize the tetrahedral intermediate in the amide bond formation (Figure 5), and a macrocycle to entropically favor the regeneration (oxidation) of the Se-Se bond [58]. This organocatalyst was used in the synthesis of various Fmoc-dipeptides under very mild conditions (23 (5 mol%), PBu3 (1.0 equiv.), 4 Å MS, MeCN, rt). This methodology was ultimately used in solid phase peptide synthesis (SPPS) for the synthesis of the pentapeptide Fmoc-FEKAG-NH2.
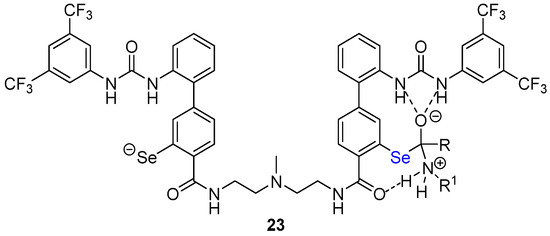
Figure 5. Urea-stabilized tetrahedral intermediate using organocatalyst 23.
The major drawbacks of the diselenide catalyst 23 are its complexity and long/tedious synthesis (10 steps). In 2022, Arora developed a simpler ‘monomeric’ catalyst 24, which still incorporates a urea moiety and a remote tertiary amine [59]. Regarding the nucleophilic phosphorus partner, phosphetane oxide 25 is used as a co-catalyst (10 mol%) and the phosphine is generated in situ in the presence of an excess of PhSiH3 (2 equiv.) (Scheme 19). No dehydrating reagent (molecular sieves) is necessary under these conditions, which makes the procedure much easier, especially in SPPS.
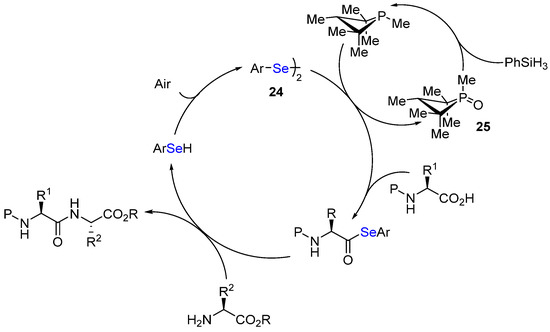
Scheme 19. The use of phosphetane oxide 25 as a phosphine precursor in 24-catalyzed amidation reactions.
Taking a different approach, though one that still requires the presence of a stoichiometric oxidant, Nguyen described tropone organocatalyst 26 [60]. The tropone 26, in the presence of oxalyl chloride (1.2 equiv.), generates 1,1-dichlorocycloheptatriene 27 and is able to react with and activate the carboxylic acid partner (Scheme 20). Such a compound can directly react with the amine to form the amide, or indirectly react through the transient formation of acyl chloride or anhydride. This strategy has been mainly used in the synthesis of esters (and lactones), but also in the synthesis of amides. The scope of this method nevertheless rather restricted (five examples in a 39–83% yield) when using dialkylamines.
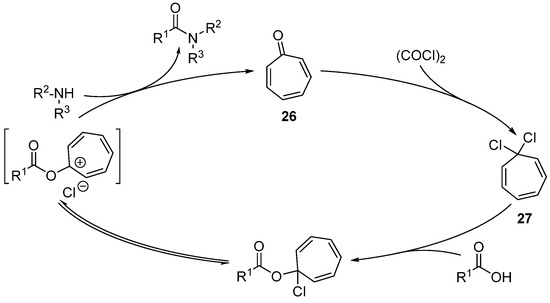
Scheme 20. Tropone-organocatalyzed amidation reactions.
Huy used sub-stoichiometric amounts of trichlorotriazine (TCT) in amidation reactions [61]. TCT is a well-known peptide coupling reagent usually used in stoichiometric amounts. In the presence of a catalytic amount of N-formylpyrrolidine (FPyr), the amount of TCT could be reduced to 40 mol%. The scope of this method is quite large (27 examples) and it exhibits good tolerance of functional groups (Scheme 21). The use of the three chlorine atoms available in TCT allows good atom economy.
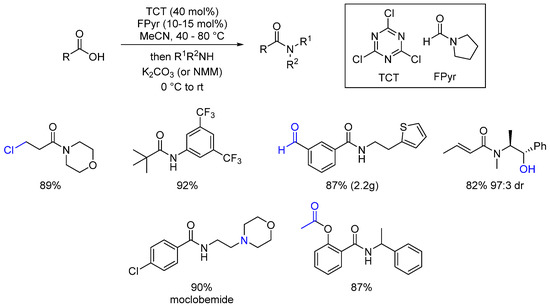
Scheme 21. Substoichiometric amounts of TCT in direct amidation reactions.
5. Metal-Catalyzed Reactions
Although the use of metals (especially titanium salts) in direct amidation reactions has been known since the 1970s, these methodologies were limited by high temperatures, relatively high catalytic loadings and limited scope. In 2012, two groundbreaking papers independently published by the Aldolfsson and Williams groups described the use of group(IV) metal salts, using zirconium and titanium salts, respectively, to allow amidification under milder conditions and with greater scope [62][63]. The mechanism of the zirconium-catalyzed reactions was then studied in a work combining kinetics, NMR and DFT experiments (Scheme 22) [64]. The dinuclear zirconium complex 28 is the actual catalytic species that is able to react one carboxylate carbon with the amine to produce the tetrahedral intermediate 29. After deprotonation (by the intervention of a second amine equivalent), the rate-determining step (with a barrier of 10.9 kcal/mol) is the cleavage of the C–O bond to produce 30, which, in turn, releases the amide bond (accompanied by a water and amine molecule) and regenerates the catalytic species 28.
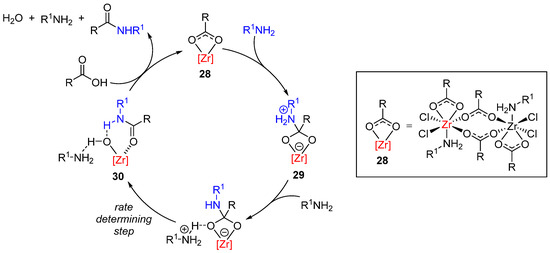
Scheme 22. Mechanistic rationale in Zr-catalyzed amidations.
In 2015, Aldolfsson was also able to develop hafnium-catalyzed reactions using 10 mol% of Cp2HfCl2, enabling reactions at room temperature with short reaction times (90 min) [65]. This method showed great scope (>25 examples) and the yields were very good. However, the reaction is sensitive to steric hindrance at the carboxylic acid partner (no reaction with Boc-Ile-OH), and no reaction was observed with anilines (Scheme 23). CAN (ceric ammonium nitrate) was also reported to be an efficient catalyst (2 mol% loading) under microwave irradiation [18].
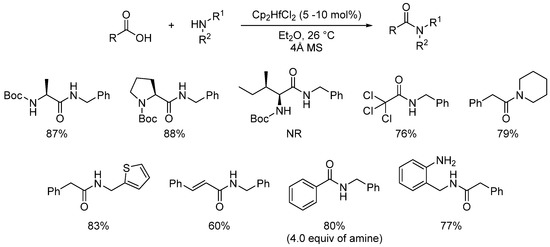
Scheme 23. Cp2HfCl2-catalyzed direct amidation reactions; NR = no reaction.
6. Miscellaneous
Kumar has described the use of substoichiometric (50 mol%) quantities of KPF6 in the amidation (and esterification) of carboxylic acids in the absence of solvent at 130 °C (14 examples) [66].
References
- Greenberg, A.; Breneman, C.M.; Liebman, J.F. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science. Wiley-Interscience: New York, NY, USA, 2000.
- Rajput, P.; Sharma, A. Synthesis and biological importance of amide analogues. J. Pharmacol. Med. Chem. 2018, 2, 22.
- Carey, J.S.; Laffan, D.; Thomsonc, C.; Williams, M.T. Analysis of the reactions used for the preparation of drug candidate molecules. Org. Biomol. Chem. 2006, 4, 2337.
- Brown, D.G.; Boström, J. Analysis of past and present synthetic methodologies on medicinal chemistry: Where have all the new reactions gone? J. Med. Chem. 2016, 59, 4443.
- De Figueiredo, R.M.; Suppo, J.-S.; Campagne, J.-M. Nonclassical routes for amide bond formation. Chem. Rev. 2016, 116, 12029.
- Ojeda-Porras, A.; Gamba-Sánchez, D. Recent developments in amide synthesis using nonactivated starting materials. J. Org. Chem. 2016, 81, 11548.
- Lundberg, H.; Tinnis, F.; Selander, N.; Adolfsson, H. Catalytic amide formation from non-activated carboxylic acids and amines. Chem. Soc. Rev. 2014, 43, 2714.
- Sabatini, M.T.; Boulton, L.T.; Sneddon, H.F.; Sheppard, T.D. A green chemistry perspective on catalytic amide bond formation. Nat. Catal. 2019, 2, 10.
- Wang, X. Challenges and outlook for catalytic direct amidation reactions. Nat. Catal. 2019, 2, 98.
- Todorovic, M.; Perrin, D.M. Recent developments in catalytic amide bond formation. Pept. Sci. 2020, 112, e24210.
- Massolo, E.; Pirola, M.; Benaglia, M. Amide bond formation strategies: Latest advances on a dateless transformation. Eur. J. Org. Chem. 2020, 2020, 4641–4645.
- Santos, A.S.; Silva, A.M.S.; Marques, M.M.B. Sustainable amidation reactions—Recent advances. Eur. J. Org. Chem. 2020, 2020, 2501.
- Cossy, J.; Pale-Grosdemange, C. A convenient synthesis of amides from carboxylic acids and primary amines. Tetrahedron Lett. 1989, 30, 2771.
- Arnold, K.; Davies, B.; Giles, R.; Grosjean, C.; Smith, G.; Whiting, A. To catalyze or not to catalyze? Insight into direct amide bond formation from amines and carboxylic acids under thermal and catalyzed conditions. Adv. Synth. Catal. 2006, 348, 813.
- Charville, H.; Jackson, D.A.; Hodges, G.; Whiting, A.; Wilson, M.R. The uncatalyzed direct amide formation reaction—Mechanism studies and the key role of carboxylic acid H-bonding. Eur. J. Org. Chem. 2011, 2011, 5981.
- Unpublished result from this laboratory.
- Perreux, L.; Loupy, A.; Volatron, F. Solvent-free preparation of amides from acids and primary amines under microwave irradiation. Tetrahedron 2002, 58, 2155.
- Zarecki, A.P.; Kolanowski, J.L.; Markiewicz, W.T. Microwave-assisted catalytic method for a green synthesis of amides directly from amines and carboxylic acids. Molecules 2020, 25, 1761.
- Magano, J. Large-scale amidations in process chemistry: Practical considerations for reagent selection and reaction execution. Org. Process Res. Dev. 2022, 26, 1562.
- El Dine, T.M.; Erb, W.; Berhault, Y.; Rouden, J.; Blanchet, J. Catalytic chemical amide synthesis at room temperature: One more step toward peptide synthesis. J. Org. Chem. 2015, 80, 4532.
- Hall, D.G. Boronic acid catalysis. Chem. Soc. Rev. 2019, 48, 3475.
- Wang, K.; Lu, Y.; Ishihara, K. The ortho-substituent on 2,4-bis(trifluoromethyl)phenylboronic acid catalyzed dehydrative condensation between carboxylic acids and amines. Chem. Commun. 2018, 54, 5410.
- Al-Zoubi, R.M.; Al-Jammal, W.K.; McDonald, R. Regioselective synthesis of ortho-iodobiphenylboronic acid derivatives: A superior catalyst for carboxylic acid activation. New J. Chem. 2020, 44, 3612.
- Ishihara, K.; Lu, Y. Boronic acid–DMAPO cooperative catalysis for dehydrative condensation between carboxylic acids and amines. Chem. Sci. 2016, 7, 1276.
- Lu, Y.; Wang, K.; Ishihara, K. Design of boronic acid-base complexes as reusable homogeneous catalysts in dehydrative condensations between carboxylic acids and amines. Asian J. Org. Chem. 2017, 6, 1191.
- Khaldoun, K.; Safer, A.; Saidi-Besbes, S.; Carboni, B.; Le Guével, R.; Carreaux, F. An efficient solvent-free microwave-assisted synthesis of cinnamamides by amidation reaction using phenylboronic acid/Lewis base Co-catalytic system. Synthesis 2019, 51, 3891.
- Arkhipenko, S.; Sabatini, M.T.; Batsanov, A.S.; Karaluka, V.; Sheppard, T.D.; Rzepa, H.S.; Whiting, A. Mechanistic insights into boron-catalysed direct amidation reactions. Chem. Sci. 2018, 9, 1058.
- Noda, H.; Furutachi, M.; Asada, Y.; Shibasaki, M.; Kumagai, N. Unique physicochemical and catalytic properties dictated by the B3NO2 ring system. Nat. Chem. 2017, 9, 571.
- Liu, Z.; Noda, H.; Shibasaki, M.; Kumagai, N. Catalytic oligopeptide synthesis. Org. Lett. 2018, 20, 612.
- Opie, C.R.; Noda, H.; Shibasaki, M.; Kumagai, N. All non-carbon B3NO2 exotic heterocycles: Synthesis, dynamics, and catalysis. Chem. Eur. J. 2019, 25, 4648.
- Noda, H.; Asada, Y.; Shibasaki, M.; Kumagai, N. Neighboring protonation unveils Lewis acidity in the B3NO2 heterocycle. J. Am. Chem. Soc. 2019, 141, 1546.
- Sawant, D.N.; Bagal, D.B.; Ogawa, S.; Selvam, K.; Saito, S. Diboron-catalyzed dehydrative amidation of aromatic carboxylic acids with amines. Org. Lett. 2018, 20, 4397.
- Shimada, N.; Hirata, M.; Koshizuka, M.; Ohse, N.; Kaito, R.; Makino, K. Diboronic acid anhydrides as effective catalysts for the hydroxy-directed dehydrative amidation of carboxylic acids. Org. Lett. 2019, 21, 4303.
- Koshizuka, M.; Makino, K.; Shimada, N. Diboronic acid anhydride-catalyzed direct peptide bond formation enabled by hydroxy-directed dehydrative condensation. Org. Lett. 2020, 22, 8658.
- Shimada, N.; Takahashi, N.; Ohse, N.; Koshizuka, M.; Makino, K. Synthesis of Weinreb amides using diboronic acid anhydride-catalyzed dehydrative amidation of carboxylic acids. Chem. Commun. 2020, 56, 13145.
- Shimada, N.; Ohse, N.; Takahashi, N.; Urata, S.; Koshizuka, M.; Makino, K. Direct synthesis of N-protected serine- and threonine-derived Weinreb amides via diboronic acid anhydride-catalyzed dehydrative amidation: Application to the concise synthesis of Garner’s aldehyde. Synlett 2021, 32, 1024.
- Michigami, K.; Sakaguchi, T.; Takemoto, Y. Catalytic dehydrative peptide synthesis with gem-diboronic acids. ACS Catal. 2020, 10, 683.
- Yohda, M.; Yamamoto, Y. Enantioselective addition of arylboronic acids to methyl 2-formylbenzoates by using a ruthenium/Me-BIPAM catalyst for synthesis of chiral 3-aryl-isobenzofuranones. Org. Biomol. Chem. 2015, 13, 10874.
- Sabatini, M.T.; Boulton, L.T.; Sheppard, T.D. Borate esters: Simple catalysts for the sustainable synthesis of complex amides. Sci. Adv. 2017, 3, e1701028.
- Sabatini, M.T.; Karaluka, V.; Lanigan, R.M.; Boulton, L.T.; Badland, M.; Sheppard, T.D. Protecting-group-free amidation of amino acids using Lewis acid catalysts. Chem. Eur. J. 2018, 24, 7033.
- Coomber, C.E.; Laserna, V.; Martin, L.T.; Smith, P.D.; Hailes, H.C.; Porter, M.J.; Sheppard, T.D. Catalytic direct amidations in tert-butyl acetate using B(OCH2CF3)3. Org. Biomol. Chem. 2019, 17, 6465.
- Mylavarapu, R.K.; GCM, K.; Kolla, N.; Veeramalla, R.; Koilkonda, P.; Bhattacharya, A.; Bandichhor, R. Boric acid catalyzed amidation in the synthesis of active pharmaceutical ingredients. Org. Proc. Res. Dev. 2007, 11, 1065.
- Shinde, G.B.; Niphade, N.C.; Deshmukh, S.P.; Toche, R.B.; Mathad, V.T. Industrial application of the Forster reaction: Novel one-pot synthesis of cinacalcet hydrochloride, a calcimimetic agent. Org. Proc. Res. Dev. 2011, 15, 455.
- Tang, P.W. Boric acid catalyzed formation from carboxylic acids and amines: N-Benzyl-4-phenylbutyramide. Org. Synth. 2012, 89, 432.
- Janvier, M.; Moebs-Sanchez, S.; Popowycz, F. Bio-based amides from renewable isosorbide by a direct and atom-economic boric acid amidation methodology. Eur. J. Org. Chem. 2016, 2016, 2308–2318.
- Chan, T.H.; Wong, L.T.L. Silicon tetrachloride as a coupling reagent for amide formation. J. Org. Chem. 1969, 34, 2766.
- Chan, T.H.; Wong, L.T.L. Evaluation of acyloxysilane as acylating agent for peptide synthesis. J. Org. Chem. 1971, 36, 850.
- Davies, J.J.; Braddock, D.C.; Lickiss, P.D. Silicon compounds as stoichiometric coupling reagents for direct amidation. Org. Biomol. Chem. 2021, 19, 6746.
- For the recent use of stoichiometric amount of silicon reagent in the presence of a co-catalytic amount of another silicon reagent in peptide synthesis, see: Muramatsu, W.; Manthena, C.; Nakashima, E.; Yamamoto, H. Peptide bond-forming reaction via amino acid silyl esters: New catalytic reactivity of an aminosilane. ACS Catal. 2020, 10, 9594.
- Lainer, T.; Czerny, F.; Haas, M. Solvent-free amide bond formation using a variety of methoxysilanes as coupling agent. Org. Biomol. Chem. 2022, 20, 3717.
- Ojeda-Porras, A.; Hernández-Santana, A.; Gamba-Sánchez, D. Direct amidation of carboxylic acids with amines under microwave irradiation using silica gel as a solid support. Green Chem. 2015, 17, 3157.
- Lenstra, D.C.; Rutjes, F.P.J.T.; Mecinović, J. Triphenylphosphine-catalysed amide bond formation between carboxylic acids and amines. Chem. Commun. 2014, 50, 5763.
- Hamstra, D.F.J.; Lenstra, D.C.; Koenders, T.J.; Rutjes, F.P.J.T.; Mecinović, J. Poly(methylhydrosiloxane) as a green reducing agent in organophosphorus-catalysed amide bond formation. Org. Biomol. Chem. 2017, 15, 6426.
- Ren, J.-W.; Tong, M.-N.; Zhao, Y.-F.; Ni, F. Synthesis of dipeptide, amide, and ester without racemization by oxalyl chloride and catalytic triphenylphosphine oxide. Org. Lett. 2021, 23, 7497.
- Movahed, F.S.; Sawant, D.N.; Bagal, D.B.; Saito, S. Tris(o-phenylenedioxy)cyclotriphosphazene as a promoter for the formation of amide bonds between aromatic acids and amines. Synthesis 2020, 52, 3253.
- Liebeskind, L.S.; Gangireddy, P.; Lindale, M.G. Benzoisothiazolone organo/copper-cocatalyzed redox dehydrative construction of amides and peptides from carboxylic acids using (EtO)3P as the reductant and O2 in air as the terminal oxidant. J. Am. Chem. Soc. 2016, 138, 6715.
- Akondi, S.M.; Gangireddy, P.; Pickel, T.C.; Liebeskind, L.S. Aerobic, diselenide-catalyzed redox dehydration: Amides and peptides. Org. Lett. 2018, 20, 538.
- Satishkumar, S.; Panigrahi, N.R.; Arora, P.S. Rational design of an organocatalyst for peptide bond formation. J. Am. Chem. Soc. 2019, 141, 15977.
- Panigrahi, N.R.; Arora, P.S. Two-component redox organocatalyst for peptide bond formation. J. Am. Chem. Soc. 2022, 144, 3637.
- Nguyen, T.V.; Lyons, D.J.M. A novel aromatic carbocation-based coupling reagent for esterification and amidation reactions. Chem. Commun. 2015, 51, 3131.
- Huy, P.H.; Mbouhom, C. Formamide catalyzed activation of carboxylic acids—Versatile and cost-efficient amidation and esterification. Chem. Sci. 2019, 10, 7399.
- Lundberg, H.; Tinnis, F.; Adolfsson, H. Direct amide coupling of non-activated carboxylic acids and amines catalysed by zirconium(IV) chloride. Chem. Eur. J. 2012, 18, 3822.
- Allen, C.L.; Chhatwal, A.R.; Williams, J.M.J. Direct amide formation from unactivated carboxylic acids and amines. Chem. Commun. 2012, 48, 666.
- Lundberg, H.; Tinnis, F.; Zhang, J.; Algarra, A.G.; Himo, F.; Adolfsson, H. Mechanistic elucidation of zirconium-catalyzed direct amidation. J. Am. Chem. Soc. 2017, 139, 2286.
- Lundberg, H.; Adolfsson, H. Hafnium-catalyzed direct amide formation at room temperature. ACS Catal. 2015, 5, 3271.
- Shinde, V.N.; Kumar, A. KPF6-mediated rsterification and amidation of carboxylic acids. J. Org. Chem. 2022, 87, 2651.
- Großmann, L.M.; Beier, V.; Duttenhofer, L.; Lennartz, L.; Opatz, T. An iodide-mediated anodic amide coupling. Chem. Eur. J. 2022, 28, e202201768.
- Nagahara, S.; Okada, Y.; Kitano, Y.; Chiba, K. Biphasic electrochemical peptide synthesis. Chem. Sci. 2021, 12, 12911.
More
Information
Subjects:
Chemistry, Organic
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
11.6K
Entry Collection:
Organic Synthesis
Revisions:
2 times
(View History)
Update Date:
22 Feb 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No