Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nobumasa Takasugi | -- | 1680 | 2023-02-20 11:56:17 | | | |
| 2 | Jessie Wu | + 6 word(s) | 1686 | 2023-02-21 03:55:55 | | | | |
| 3 | Jessie Wu | + 10 word(s) | 1696 | 2023-02-21 03:59:45 | | | | |
| 4 | Jessie Wu | + 2 word(s) | 1698 | 2023-02-21 04:01:34 | | | | |
| 5 | Jessie Wu | + 12 word(s) | 1710 | 2023-02-21 04:04:40 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Takasugi, N.; Komai, M.; Kaneshiro, N.; Ikeda, A.; Kamikubo, Y.; Uehara, T. Impact of C99 on Alzheimer’s Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/41430 (accessed on 23 July 2026).
Takasugi N, Komai M, Kaneshiro N, Ikeda A, Kamikubo Y, Uehara T. Impact of C99 on Alzheimer’s Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/41430. Accessed July 23, 2026.
Takasugi, Nobumasa, Masato Komai, Nanaka Kaneshiro, Atsuya Ikeda, Yuji Kamikubo, Takashi Uehara. "Impact of C99 on Alzheimer’s Disease" Encyclopedia, https://encyclopedia.pub/entry/41430 (accessed July 23, 2026).
Takasugi, N., Komai, M., Kaneshiro, N., Ikeda, A., Kamikubo, Y., & Uehara, T. (2023, February 20). Impact of C99 on Alzheimer’s Disease. In Encyclopedia. https://encyclopedia.pub/entry/41430
Takasugi, Nobumasa, et al. "Impact of C99 on Alzheimer’s Disease." Encyclopedia. Web. 20 February, 2023.
Copy Citation
Amyloid beta (Aβ) is produced from a type-I transmembrane protein, amyloid beta precursor protein (APP). One of the APP metabolites, the 99-amino acids C-terminal fragment (C99, also called βCTF), is a direct precursor of Aβ and accumulates in the Alzheimer’s disease (AD) patient’s brain to demonstrate toxicity independent of Aβ. Conventional drug discovery strategies have focused on Aβ toxicity on the “outside” of the neuron, but C99 accumulation might explain the toxicity on the “inside” of the neuron, which was overlooked in the hypothesis. Furthermore, the common region of C99 and Aβ is a promising target for multifunctional AD drugs.
Alzheimer’s disease
C99
patient
1. 99-Amino Acids C-Terminal Fragment Accumulation in Alzheimer’s Disease Pathology and Other Neurodegenerative Diseases
99-Amino acids C-terminal fragment has attracted attention as an “intracellular” pathology promoter in the neurons. C99 is normally produced at low levels, but when amounts are increased, such as by activation of BACE1, it is not readily degraded and accumulates in the brain. This type of property is important in the etiologic factors in neurodegenerative diseases that progress with aging. This chapter outlines C99 accumulation and the methods for its analysis in AD and other neurodegenerative diseases and their models.
Alzheimer’s disease (AD) patients, particularly familial Alzheimer’s disease (FAD) patients with mutations in amyloid beta precursor protein (APP) or Presenilin 1, showed a significant accumulation of C99 [1]. Similar accumulation of C99 has been reported in AD cell models in which APP and Presenilin 1 mutations of FAD were artificially introduced into iPS cells, suggesting that this is a common AD pathology [2][3]. Many of these mutations increase the production ratio of Aβ42, seemingly contradicting the accumulation of the substrate, C99. However, the increase in Aβ42 can be understood as a change in γ-secretase cleavage site. APP mutations in the proximal site of γ-secretase cleavage enhance the Aβ42 generation rate, but some of them tend to increase the APP-CTF stability [4]. It was also reported that FAD mutations in Presenilin1 shift the cleavage site toward Aβ42 production due to reduced secretase activity [5], or impaired proteolysis in autolysosome [6]. The commonality shown by these lines of evidence is that C99 is once produced, less likely to be degraded. Furthermore, as will be discussed in later chapters, there is also a vicious cycle in which C99 accumulation impairs the endolysosomal and autolysosomal pathways, which impairs the metabolism of C99 itself. These findings suggest that the pathogenic mechanism of AD has a cross link to the accumulation of C99.
Would C99 accumulation be associated with pathology? As C99 contains the amyloid beta (Aβ) sequence, histological differentiation of C99 and Aβ is difficult. Applying β1 cleavage site-specific (antibodies detect both Aβ and C99), and APP C-terminal antibodies to the proximal ligation assay (PLA) enables the analysis of the precise localization of C99. Using this technique, it was observed that C99 accumulated in AD brains in regions susceptible to AD, such as CA1 and the dentate gyrus, and that this correlated with pathological conditions such as cognitive decline [7]. Similar results were obtained with standard histological analyses using model mice [8]. Thus, the accumulation of C99 occurs in AD model mice, but most AD model mice have the Swedish APP mutation, which promotes the production of C99 and makes its accumulation artificial. Furthermore, additional mutations that accelerate Aβ aggregation are added in many models, making it difficult to distinguish between C99 and Aβ aggregation toxicity. Efforts are being made to distinguish them, including using γ-secretase inhibitors, which increase C99 while suppressing Aβ production, and β-secretase inhibitors, which decrease both Aβ and C99. Additionally, immunostaining using a combination of specific antibodies, the PLA analysis [7], ELISA [9], and immunoblotting [10] are used to determine the C99 specific distribution and quantification. These analyses showed that C99 accumulation precedes Aβ deposition in model mice and mediates Aβ independent intracellular pathologies [8][9][10][11]. However, limitations of the analysis using standard AD model mice may need to be addressed. For C99-specific analysis, models such as the A7 mouse [10][12], in which the Aβ deposition is slow, and the model without the Swedish mutation [13] may be helpful.
Additionally, other neurodegenerative diseases accumulate C99 and affect pathology, and these conditions can also provide important clues. Down syndrome (DS) is caused by trisomy of chromosome 21 and develops AD at a high rate and early age, mainly due to the triplication of the APP gene. Therefore, the DS can be a pathological model of AD. As discussed in later chapters, the initial pathology of DS and AD includes endolysosomal impairments represented by enlarged endosomes [14], which has been linked to the accumulation of C99.
Niemann-Pick disease type C (NPC) is an autosomal recessive neurodegenerative disease associated with mutations in the NPC1 or NPC2 genes of human chromosome 18, which code for lysosomal lipid transport proteins. Dysfunction of this transport protein causes the accumulation of free cholesterol in lysosomes throughout the body and accumulation of sphingolipids, especially in cranial nerve cells. Accumulation of APP-CTFs, including C99, and changes in their localization occur in model cells of genetic and drug-induced NPC [15][16], which shows similar pathology with AD such as NFT and Aβ accumulations [17][18][19][20]. Interestingly, in Purkinje cells in the brains of NPC patients with, C99, but not Aβ42, it accumulates in early endosomes and induces a redistribution of lysosomal proteins, such as cathepsin D, to the endosomes. Additionally, enlargement of Rab5-positive early endosomes has been observed in the brains of NPC patients [15], similar to AD and DS pathology. The extent of the contribution of C99 to NPC pathogenesis needs to be verified in the future. However, since NPC is a disease associated with abnormal lipid and cholesterol metabolism and accumulation, these impairments may affect C99 metabolism. Furthermore, these lines of evidence also suggest that accumulated C99 itself may be involved in disturbances of endolysosomes and lipid metabolism, creating a vicious cycle.
2. Structure and Metabolism of 99-Amino Acids C-Terminal Fragment
To consider the unique properties of C99, a comparison with C83, would be helpful. To repeat from the previous chapter, compared to C83, C99 is challenging to produce, but difficult to break once formed. What produces the characteristic properties of C99? Here, researchers focus on the function of the Aβ N-terminal region (Aβ-Nt; Aβ 1–16aa), which is missing in C83. The sequence differences in this region between humans and mice are related to aggregation and exerted toxicity [21], as well as specific in APP among other family proteins such as APLP1 and APLP2 [22], and their involvement in AD pathogenesis has attracted attention [23].
Genetic evidence suggests the importance of this region. The first is the Swedish mutation, which is present just before the β1 cleavage site and increases the production of C99 and Aβ. The second is the English (H7R) and Tottori (D8N) FAD mutations, which affect the formation of Aβ fibril [24] and oligomer [25], that are deeply involved in AD pathogenesis [26]. Furthermore, the presence of the Uppsala mutation (deletion of Aβ_19F-24V), in which the extracellular domain of C99 is deleted, leaving Aβ-Nt, which is supposed to induce AD, also suggests a requirement for Aβ-Nt in the development of AD [27]. Biochemically, the Uppsala mutation is associated with increased Aβ production and aggregation, as well as C99 accumulation. Although the Uppsala mutation has few patients and may need further validation, these mutation carriers show a younger onset age than other FAD mutations in their forties. Their pathological progression is reported to be more rapid.
If this is the case, then how does Aβ-Nt affect the C99 structure? Solution NMR measurements using zwitterionic lipid bicelles and micelles have provided the structural information on the extracellular domain of C99 [28][29]. In those analyses, the N-terminal region (1–15) forms a disordered structure, and the following seven amino acid residues form an N-helix structure that binds to the surface of membrane components and binds to the transmembrane region via a flexible five amino acid loop. In contrast to transmembrane domain to N-helix structure is predicted to have cholesterol-binding properties in a cooperative manner [28], the structure of the Aβ-Nt is disordered, which might determine the properties through interactions by binding to other factors. It has also been suggested that, depending on membrane thickness conditions, Aβ-Nt adopt a β-hairpin structure lifted on membrane surface and function as a starting point for Aβ aggregation in concert with other extracellular domains [29] (Figure 1).
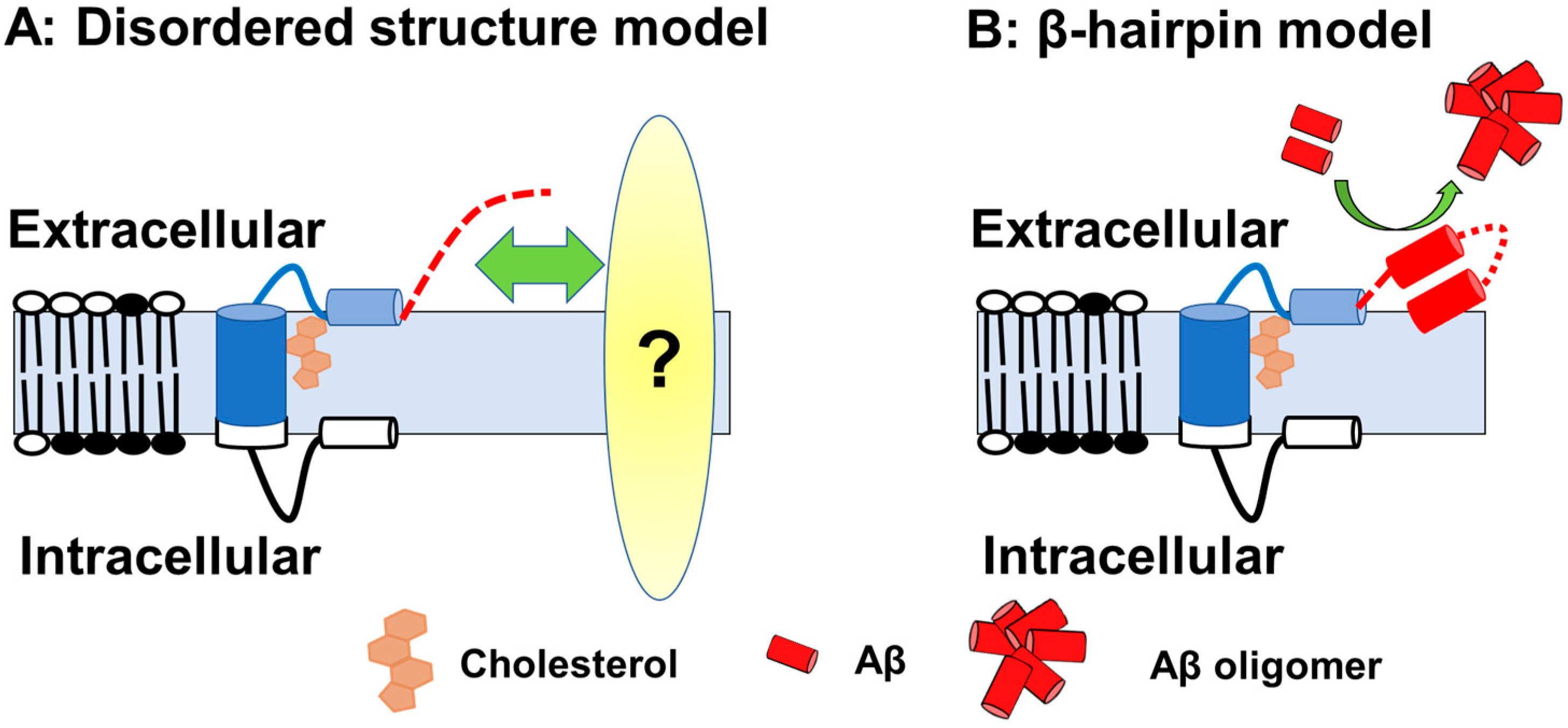
Figure 1. A schematic of the C99 structure. (A) Model with Aβ-Nt as the disordered structure; Aβ-Nt may bind to its partner molecule and exert specific functions. Red dotted line represented N-terminal structure. (B) Model assuming Aβ-Nt forms a β-hairpin. The β-hairpin structure formed may promote Aβ aggregation.
So how does Aβ-Nt affect C99 properties? C99 undergoes several metabolic pathways, one of which is the γ-secretase pathway. Although, Aβ production is crossly correlated with the C99 level, C99 is not a good substrate for γ-secretase compared to C83 and other γ-secretase substrates, Notch. The γ-secretase activity analysis in vitro [30] or cell-based reporters [31] indicates that C83 and Notch are cleaved more easily than C99. Two reasons are postulated: the first is the difference in the length of the extracellular region, and the second is that this extracellular region has an inhibitory function on γ-secretase cleavage. On that basis, a drosophila reporter system has shown that the shorter the extracellular domain, the more efficiently the substrate is cleaved by γ-secretase substrates [32]. Furthermore, the Aβ-Nt sequence has an inhibitory structure for γ-secretase cleavage [30], and C99 accumulation has been predicted to saturate the substrate binding site of γ-secretase, thereby decreasing its activity [33].
The other major metabolic pathway is degradation in the autolysosomal pathway. C99 and other APP-CTFs have been reported to be transported to lysosomes and subjected to degradation by β-cathepsin [34]. APP-CTF in the endocytic pathway interacts with a complex formed by AP2 (adaptor-related protein complex 2) and, one of the AD risk factors, PICALM (phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia). This complex interacts with autophagy protein LC3 (microtubule-associated protein one light chain 3) to transport C99 to the autolysosomal pathway (70, 71). Autolysosomal pathways are affected by the efficiency of vesicular transport by endosomes, and disturbances in both pathways have been reported in AD and other neurodegenerative diseases [35]. As will be discussed in more detail in later chapters, accumulated C99 may contribute to the impairment of these pathways and promote a vicious cycle of AD progression.
References
- Pera, M.; Alcolea, D.; Sanchez-Valle, R.; Guardia-Laguarta, C.; Colom-Cadena, M.; Badiola, N.; Suarez-Calvet, M.; Llado, A.; Barrera-Ocampo, A.A.; Sepulveda-Falla, D.; et al. Distinct patterns of APP processing in the CNS in autosomal-dominant and sporadic Alzheimer disease. Acta Neuropathol. 2013, 125, 201–213.
- Hung, C.O.Y.; Livesey, F.J. Altered γ-Secretase Processing of APP Disrupts Lysosome and Autophagosome Function in Monogenic Alzheimer’s Disease. Cell Rep. 2018, 25, 3647–3660 e3642.
- Kwart, D.; Gregg, A.; Scheckel, C.; Murphy, E.A.; Paquet, D.; Duffield, M.; Fak, J.; Olsen, O.; Darnell, R.B.; Tessier-Lavigne, M. A Large Panel of Isogenic APP and PSEN1 Mutant Human iPSC Neurons Reveals Shared Endosomal Abnormalities Mediated by APP β-CTFs, Not Aβ. Neuron 2019, 104, 256–270.e255.
- De Jonghe, C. Pathogenic APP mutations near the γ-secretase cleavage site differentially affect Aβ secretion and APP C-terminal fragment stability. Hum. Mol. Genet. 2001, 10, 1665–1671.
- Szaruga, M.; Munteanu, B.; Lismont, S.; Veugelen, S.; Horre, K.; Mercken, M.; Saido, T.C.; Ryan, N.S.; De Vos, T.; Savvides, S.N.; et al. Alzheimer’s-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions. Cell 2017, 170, 443–456.e14.
- Lee, J.-H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal Proteolysis and Autophagy Require Presenilin 1 and Are Disrupted by Alzheimer-Related PS1 Mutations. Cell 2010, 141, 1146–1158.
- Pulina, M.V.; Hopkins, M.; Haroutunian, V.; Greengard, P.; Bustos, V. C99 selectively accumulates in vulnerable neurons in Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 273–282.
- Lauritzen, I.; Pardossi-Piquard, R.; Bauer, C.; Brigham, E.; Abraham, J.D.; Ranaldi, S.; Fraser, P.; St-George-Hyslop, P.; Le Thuc, O.; Espin, V.; et al. The β-secretase-derived C-terminal fragment of βAPP, C99, but not Aβ, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J. Neurosci. 2012, 32, 16243–16255.
- Lauritzen, I.; Pardossi-Piquard, R.; Bourgeois, A.; Pagnotta, S.; Biferi, M.G.; Barkats, M.; Lacor, P.; Klein, W.; Bauer, C.; Checler, F. Intraneuronal aggregation of the β-CTF fragment of APP (C99) induces Aβ-independent lysosomal-autophagic pathology. Acta Neuropathol. 2016, 132, 257–276.
- Kaneshiro, N.; Komai, M.; Imaoka, R.; Ikeda, A.; Kamikubo, Y.; Saito, T.; Saido, T.C.; Tomita, T.; Hashimoto, T.; Iwatsubo, T.; et al. Lipid flippase dysfunction as a therapeutic target for endosomal anomalies in Alzheimer’s disease. iScience 2022, 25, 103869.
- Kaur, G.; Pawlik, M.; Gandy, S.E.; Ehrlich, M.E.; Smiley, J.F.; Levy, E. Lysosomal dysfunction in the brain of a mouse model with intraneuronal accumulation of carboxyl terminal fragments of the amyloid precursor protein. Mol. Psychiatry 2017, 22, 981–989.
- Yamada, K.; Yabuki, C.; Seubert, P.; Schenk, D.; Hori, Y.; Ohtsuki, S.; Terasaki, T.; Hashimoto, T.; Iwatsubo, T. Aβ immunotherapy: Intracerebral sequestration of Aβ by an anti-Aβ monoclonal antibody 266 with high affinity to soluble Aβ. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 11393–11398.
- Watamura, N.; Sato, K.; Shiihashi, G.; Iwasaki, A.; Kamano, N.; Takahashi, M.; Sekiguchi, M.; Mihira, N.; Fujioka, R.; Nagata, K.; et al. An isogenic panel of App knock-in mouse models: Profiling β-secretase inhibition and endosomal abnormalities. Sci. Adv. 2022, 8, eabm6155.
- Cataldo, A.M.; Peterhoff, C.M.; Troncoso, J.C.; Gomez-Isla, T.; Hyman, B.T.; Nixon, R.A. Endocytic pathway abnormalities precede amyloid β deposition in sporadic Alzheimer’s disease and Down syndrome: Differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 2000, 157, 277–286.
- Jin, L.-W.; Maezawa, I.; Vincent, I.; Bird, T. Intracellular Accumulation of Amyloidogenic Fragments of Amyloid-β Precursor Protein in Neurons with Niemann-Pick Type C Defects Is Associated with Endosomal Abnormalities. Am. J. Pathol. 2004, 164, 975–985.
- Kosicek, M.; Malnar, M.; Goate, A.; Hecimovic, S. Cholesterol accumulation in Niemann Pick type C (NPC) model cells causes a shift in APP localization to lipid rafts. Biochem. Biophys. Res. Commun. 2010, 393, 404–409.
- Cariati, I.; Masuelli, L.; Bei, R.; Tancredi, V.; Frank, C.; D’Arcangelo, G. Neurodegeneration in Niemann–Pick Type C Disease: An Updated Review on Pharmacological and Non-Pharmacological Approaches to Counteract Brain and Cognitive Impairment. Int. J. Mol. Sci. 2021, 22, 6600.
- Suzuki, K.; Parker, C.C.; Pentchev, P.G.; Katz, D.; Ghetti, B.; D’Agostino, A.N.; Carstea, E.D. Neurofibrillary tangles in Niemann-Pick disease type C. Acta Neuropathol. 1995, 89, 227–238.
- Love, S.; Bridges, L.R.; Case, C.P. Neurofibrillary tangles in Niemann—Pick disease type C. Brain A J. Neurol. 1995, 118, 119–129.
- Yamazaki, T.; Chang, T.-Y.; Haass, C.; Ihara, Y. Accumulation and Aggregation of Amyloid β-Protein in Late Endosomes of Niemann-Pick Type C Cells. J. Biol. Chem. 2001, 276, 4454–4460.
- Lv, X.; Li, W.; Luo, Y.; Wang, D.; Zhu, C.; Huang, Z.X.; Tan, X. Exploring the differences between mouse mAβ(1-42) and human hAβ(1-42) for Alzheimer’s disease related properties and neuronal cytotoxicity. Chem. Commun. 2013, 49, 5865–5867.
- Zheng, H.; Koo, E.H. The amyloid precursor protein: Beyond amyloid. Mol. Neurodegener. 2006, 1, 5.
- Murray, B.; Sharma, B.; Belfort, G. N-Terminal Hypothesis for Alzheimer’s Disease. ACS Chem. Neurosci. 2017, 8, 432–434.
- Hori, Y.; Hashimoto, T.; Wakutani, Y.; Urakami, K.; Nakashima, K.; Condron, M.M.; Tsubuki, S.; Saido, T.C.; Teplow, D.B.; Iwatsubo, T. The Tottori (D7N) and English (H6R) familial Alzheimer disease mutations accelerate Aβ fibril formation without increasing protofibril formation. J. Biol. Chem. 2007, 282, 4916–4923.
- Ono, K.; Condron, M.M.; Teplow, D.B. Effects of the English (H6R) and Tottori (D7N) Familial Alzheimer Disease Mutations on Amyloid β-Protein Assembly and Toxicity. J. Biol. Chem. 2010, 285, 23186–23197.
- Yang, H.; Li, J.; Li, X.; Ma, L.; Hou, M.; Zhou, H.; Zhou, R. Based on molecular structures: Amyloid-β generation, clearance, toxicity and therapeutic strategies. Front. Mol. Neurosci. 2022, 15, 927530.
- Pagnon de la Vega, M.; Giedraitis, V.; Michno, W.; Kilander, L.; Guner, G.; Zielinski, M.; Lowenmark, M.; Brundin, R.; Danfors, T.; Soderberg, L.; et al. The Uppsala APP deletion causes early onset autosomal dominant Alzheimer’s disease by altering APP processing and increasing amyloid β fibril formation. Sci. Transl. Med. 2021, 13, eabc6184.
- Barrett, P.J.; Song, Y.; Van Horn, W.D.; Hustedt, E.J.; Schafer, J.M.; Hadziselimovic, A.; Beel, A.J.; Sanders, C.R. The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 2012, 336, 1168–1171.
- Pantelopulos, G.A.; Straub, J.E.; Thirumalai, D.; Sugita, Y. Structure of APP-C991-99 and implications for role of extra-membrane domains in function and oligomerization. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1698–1708.
- Funamoto, S.; Sasaki, T.; Ishihara, S.; Nobuhara, M.; Nakano, M.; Watanabe-Takahashi, M.; Saito, T.; Kakuda, N.; Miyasaka, T.; Nishikawa, K.; et al. Substrate ectodomain is critical for substrate preference and inhibition of γ-secretase. Nat. Commun. 2013, 4, 2529.
- Yan, Y.; Xu, T.-H.; Melcher, K.; Xu, H.E. Defining the minimum substrate and charge recognition model of γ-secretase. Acta Pharmacol. Sin. 2017, 38, 1412–1424.
- Struhl, G.; Adachi, A. Requirements for Presenilin-Dependent Cleavage of Notch and Other Transmembrane Proteins. Mol. Cell 2000, 6, 625–636.
- Svedružić, Ž.M.; Jengić, V.Š.; Ostojić, L. Binding of different substrate molecules at the docking site and the active site of γ-secretase can trigger toxic events in sporadic and familial Alzheimer’s disease. bioRxiv 2022, 24, 1835.
- Asai, M.; Yagishita, S.; Iwata, N.; Saido, T.C.; Ishiura, S.; Maruyama, K. An alternative metabolic pathway of amyloid precursor protein C-terminal fragments via cathepsin B in a human neuroglioma model. FASEB J. 2011, 25, 3720–3730.
- Nixon, R.A. The aging lysosome: An essential catalyst for late-onset neurodegenerative diseases. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140443.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Entry Collection:
Neurodegeneration
Revisions:
5 times
(View History)
Update Date:
21 Feb 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No