Amyloid beta (Aβ) is produced from a type-I transmembrane protein, amyloid beta precursor protein (APP). One of the APP metabolites, the 99-amino acids C-terminal fragment (C99, also called βCTF), is a direct precursor of Aβ and accumulates in the Alzheimer’s disease (AD) patient’s brain to demonstrate toxicity independent of Aβ. Conventional drug discovery strategies have focused on Aβ toxicity on the “outside” of the neuron, but C99 accumulation might explain the toxicity on the “inside” of the neuron, which was overlooked in the hypothesis. Furthermore, the common region of C99 and Aβ is a promising target for multifunctional AD drugs.
- Alzheimer’s disease
- C99
- patient
1. 99-Amino Acids C-Terminal Fragment Accumulation in Alzheimer’s Disease Pathology and Other Neurodegenerative Diseases
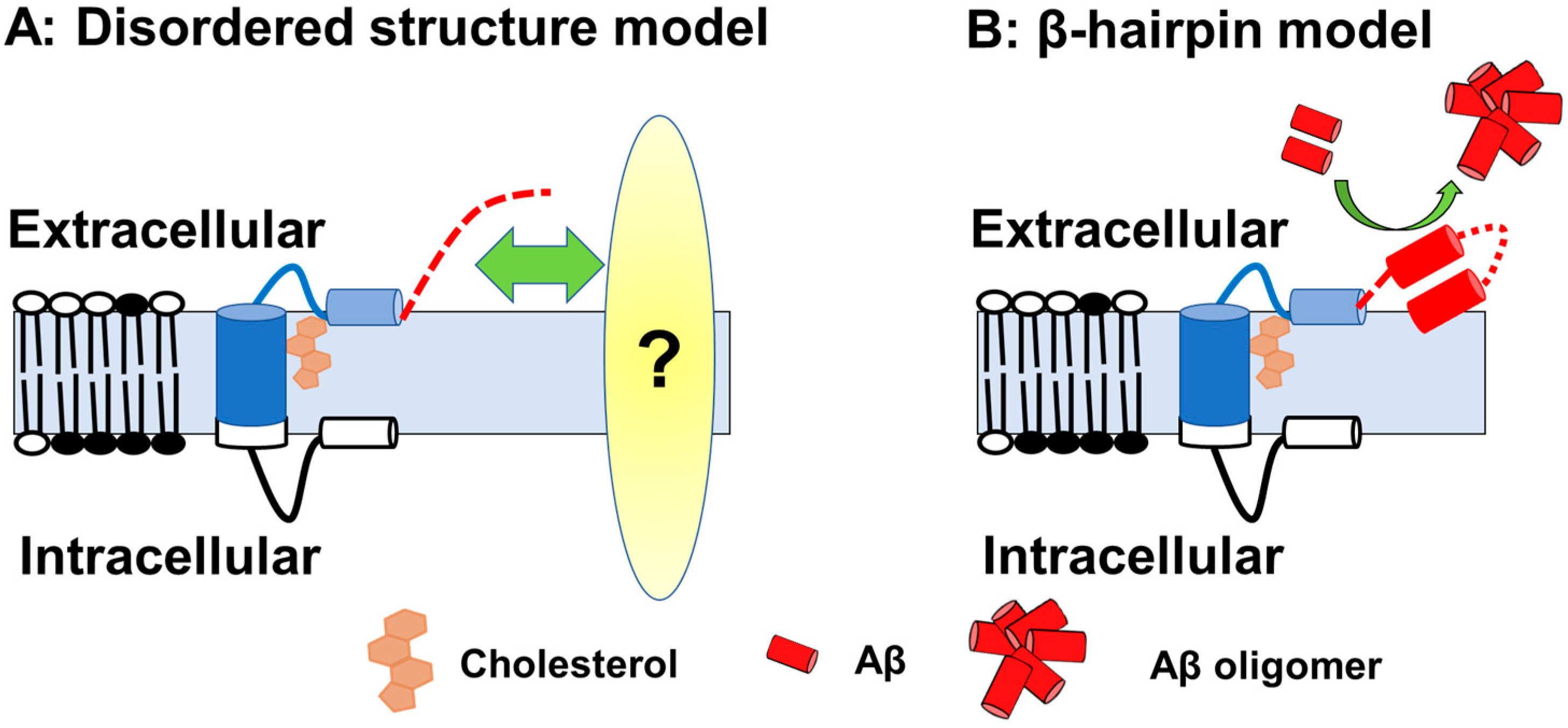
2. Structure and Metabolism of 99-Amino Acids C-Terminal Fragment
This entry is adapted from the peer-reviewed paper 10.3390/cells12030454
References
- Pera, M.; Alcolea, D.; Sanchez-Valle, R.; Guardia-Laguarta, C.; Colom-Cadena, M.; Badiola, N.; Suarez-Calvet, M.; Llado, A.; Barrera-Ocampo, A.A.; Sepulveda-Falla, D.; et al. Distinct patterns of APP processing in the CNS in autosomal-dominant and sporadic Alzheimer disease. Acta Neuropathol. 2013, 125, 201–213.
- Hung, C.O.Y.; Livesey, F.J. Altered γ-Secretase Processing of APP Disrupts Lysosome and Autophagosome Function in Monogenic Alzheimer’s Disease. Cell Rep. 2018, 25, 3647–3660 e3642.
- Kwart, D.; Gregg, A.; Scheckel, C.; Murphy, E.A.; Paquet, D.; Duffield, M.; Fak, J.; Olsen, O.; Darnell, R.B.; Tessier-Lavigne, M. A Large Panel of Isogenic APP and PSEN1 Mutant Human iPSC Neurons Reveals Shared Endosomal Abnormalities Mediated by APP β-CTFs, Not Aβ. Neuron 2019, 104, 256–270.e255.
- De Jonghe, C. Pathogenic APP mutations near the γ-secretase cleavage site differentially affect Aβ secretion and APP C-terminal fragment stability. Hum. Mol. Genet. 2001, 10, 1665–1671.
- Szaruga, M.; Munteanu, B.; Lismont, S.; Veugelen, S.; Horre, K.; Mercken, M.; Saido, T.C.; Ryan, N.S.; De Vos, T.; Savvides, S.N.; et al. Alzheimer’s-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions. Cell 2017, 170, 443–456.e14.
- Lee, J.-H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal Proteolysis and Autophagy Require Presenilin 1 and Are Disrupted by Alzheimer-Related PS1 Mutations. Cell 2010, 141, 1146–1158.
- Pulina, M.V.; Hopkins, M.; Haroutunian, V.; Greengard, P.; Bustos, V. C99 selectively accumulates in vulnerable neurons in Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 273–282.
- Lauritzen, I.; Pardossi-Piquard, R.; Bauer, C.; Brigham, E.; Abraham, J.D.; Ranaldi, S.; Fraser, P.; St-George-Hyslop, P.; Le Thuc, O.; Espin, V.; et al. The β-secretase-derived C-terminal fragment of βAPP, C99, but not Aβ, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J. Neurosci. 2012, 32, 16243–16255.
- Lauritzen, I.; Pardossi-Piquard, R.; Bourgeois, A.; Pagnotta, S.; Biferi, M.G.; Barkats, M.; Lacor, P.; Klein, W.; Bauer, C.; Checler, F. Intraneuronal aggregation of the β-CTF fragment of APP (C99) induces Aβ-independent lysosomal-autophagic pathology. Acta Neuropathol. 2016, 132, 257–276.
- Kaneshiro, N.; Komai, M.; Imaoka, R.; Ikeda, A.; Kamikubo, Y.; Saito, T.; Saido, T.C.; Tomita, T.; Hashimoto, T.; Iwatsubo, T.; et al. Lipid flippase dysfunction as a therapeutic target for endosomal anomalies in Alzheimer’s disease. iScience 2022, 25, 103869.
- Kaur, G.; Pawlik, M.; Gandy, S.E.; Ehrlich, M.E.; Smiley, J.F.; Levy, E. Lysosomal dysfunction in the brain of a mouse model with intraneuronal accumulation of carboxyl terminal fragments of the amyloid precursor protein. Mol. Psychiatry 2017, 22, 981–989.
- Yamada, K.; Yabuki, C.; Seubert, P.; Schenk, D.; Hori, Y.; Ohtsuki, S.; Terasaki, T.; Hashimoto, T.; Iwatsubo, T. Aβ immunotherapy: Intracerebral sequestration of Aβ by an anti-Aβ monoclonal antibody 266 with high affinity to soluble Aβ. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 11393–11398.
- Watamura, N.; Sato, K.; Shiihashi, G.; Iwasaki, A.; Kamano, N.; Takahashi, M.; Sekiguchi, M.; Mihira, N.; Fujioka, R.; Nagata, K.; et al. An isogenic panel of App knock-in mouse models: Profiling β-secretase inhibition and endosomal abnormalities. Sci. Adv. 2022, 8, eabm6155.
- Cataldo, A.M.; Peterhoff, C.M.; Troncoso, J.C.; Gomez-Isla, T.; Hyman, B.T.; Nixon, R.A. Endocytic pathway abnormalities precede amyloid β deposition in sporadic Alzheimer’s disease and Down syndrome: Differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 2000, 157, 277–286.
- Jin, L.-W.; Maezawa, I.; Vincent, I.; Bird, T. Intracellular Accumulation of Amyloidogenic Fragments of Amyloid-β Precursor Protein in Neurons with Niemann-Pick Type C Defects Is Associated with Endosomal Abnormalities. Am. J. Pathol. 2004, 164, 975–985.
- Kosicek, M.; Malnar, M.; Goate, A.; Hecimovic, S. Cholesterol accumulation in Niemann Pick type C (NPC) model cells causes a shift in APP localization to lipid rafts. Biochem. Biophys. Res. Commun. 2010, 393, 404–409.
- Cariati, I.; Masuelli, L.; Bei, R.; Tancredi, V.; Frank, C.; D’Arcangelo, G. Neurodegeneration in Niemann–Pick Type C Disease: An Updated Review on Pharmacological and Non-Pharmacological Approaches to Counteract Brain and Cognitive Impairment. Int. J. Mol. Sci. 2021, 22, 6600.
- Suzuki, K.; Parker, C.C.; Pentchev, P.G.; Katz, D.; Ghetti, B.; D’Agostino, A.N.; Carstea, E.D. Neurofibrillary tangles in Niemann-Pick disease type C. Acta Neuropathol. 1995, 89, 227–238.
- Love, S.; Bridges, L.R.; Case, C.P. Neurofibrillary tangles in Niemann—Pick disease type C. Brain A J. Neurol. 1995, 118, 119–129.
- Yamazaki, T.; Chang, T.-Y.; Haass, C.; Ihara, Y. Accumulation and Aggregation of Amyloid β-Protein in Late Endosomes of Niemann-Pick Type C Cells. J. Biol. Chem. 2001, 276, 4454–4460.
- Lv, X.; Li, W.; Luo, Y.; Wang, D.; Zhu, C.; Huang, Z.X.; Tan, X. Exploring the differences between mouse mAβ(1-42) and human hAβ(1-42) for Alzheimer’s disease related properties and neuronal cytotoxicity. Chem. Commun. 2013, 49, 5865–5867.
- Zheng, H.; Koo, E.H. The amyloid precursor protein: Beyond amyloid. Mol. Neurodegener. 2006, 1, 5.
- Murray, B.; Sharma, B.; Belfort, G. N-Terminal Hypothesis for Alzheimer’s Disease. ACS Chem. Neurosci. 2017, 8, 432–434.
- Hori, Y.; Hashimoto, T.; Wakutani, Y.; Urakami, K.; Nakashima, K.; Condron, M.M.; Tsubuki, S.; Saido, T.C.; Teplow, D.B.; Iwatsubo, T. The Tottori (D7N) and English (H6R) familial Alzheimer disease mutations accelerate Aβ fibril formation without increasing protofibril formation. J. Biol. Chem. 2007, 282, 4916–4923.
- Ono, K.; Condron, M.M.; Teplow, D.B. Effects of the English (H6R) and Tottori (D7N) Familial Alzheimer Disease Mutations on Amyloid β-Protein Assembly and Toxicity. J. Biol. Chem. 2010, 285, 23186–23197.
- Yang, H.; Li, J.; Li, X.; Ma, L.; Hou, M.; Zhou, H.; Zhou, R. Based on molecular structures: Amyloid-β generation, clearance, toxicity and therapeutic strategies. Front. Mol. Neurosci. 2022, 15, 927530.
- Pagnon de la Vega, M.; Giedraitis, V.; Michno, W.; Kilander, L.; Guner, G.; Zielinski, M.; Lowenmark, M.; Brundin, R.; Danfors, T.; Soderberg, L.; et al. The Uppsala APP deletion causes early onset autosomal dominant Alzheimer’s disease by altering APP processing and increasing amyloid β fibril formation. Sci. Transl. Med. 2021, 13, eabc6184.
- Barrett, P.J.; Song, Y.; Van Horn, W.D.; Hustedt, E.J.; Schafer, J.M.; Hadziselimovic, A.; Beel, A.J.; Sanders, C.R. The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 2012, 336, 1168–1171.
- Pantelopulos, G.A.; Straub, J.E.; Thirumalai, D.; Sugita, Y. Structure of APP-C991-99 and implications for role of extra-membrane domains in function and oligomerization. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1698–1708.
- Funamoto, S.; Sasaki, T.; Ishihara, S.; Nobuhara, M.; Nakano, M.; Watanabe-Takahashi, M.; Saito, T.; Kakuda, N.; Miyasaka, T.; Nishikawa, K.; et al. Substrate ectodomain is critical for substrate preference and inhibition of γ-secretase. Nat. Commun. 2013, 4, 2529.
- Yan, Y.; Xu, T.-H.; Melcher, K.; Xu, H.E. Defining the minimum substrate and charge recognition model of γ-secretase. Acta Pharmacol. Sin. 2017, 38, 1412–1424.
- Struhl, G.; Adachi, A. Requirements for Presenilin-Dependent Cleavage of Notch and Other Transmembrane Proteins. Mol. Cell 2000, 6, 625–636.
- Svedružić, Ž.M.; Jengić, V.Š.; Ostojić, L. Binding of different substrate molecules at the docking site and the active site of γ-secretase can trigger toxic events in sporadic and familial Alzheimer’s disease. bioRxiv 2022, 24, 1835.
- Asai, M.; Yagishita, S.; Iwata, N.; Saido, T.C.; Ishiura, S.; Maruyama, K. An alternative metabolic pathway of amyloid precursor protein C-terminal fragments via cathepsin B in a human neuroglioma model. FASEB J. 2011, 25, 3720–3730.
- Nixon, R.A. The aging lysosome: An essential catalyst for late-onset neurodegenerative diseases. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140443.