Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ester Blanco | -- | 2781 | 2023-02-15 12:05:07 | | | |
| 2 | Rita Xu | Meta information modification | 2781 | 2023-02-16 02:11:40 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Blanco, E.; Chocarro, L.; Fernández-Rubio, L.; Bocanegra, A.; Arasanz, H.; Echaide, M.; Garnica, M.; Piñeiro-Hermida, S.; Kochan, G.; Escors, D. Monoclonal Antibodies for the Treatment of Solid Tumors. Encyclopedia. Available online: https://encyclopedia.pub/entry/41252 (accessed on 25 June 2026).
Blanco E, Chocarro L, Fernández-Rubio L, Bocanegra A, Arasanz H, Echaide M, et al. Monoclonal Antibodies for the Treatment of Solid Tumors. Encyclopedia. Available at: https://encyclopedia.pub/entry/41252. Accessed June 25, 2026.
Blanco, Ester, Luisa Chocarro, Leticia Fernández-Rubio, Ana Bocanegra, Hugo Arasanz, Miriam Echaide, Maider Garnica, Sergio Piñeiro-Hermida, Grazyna Kochan, David Escors. "Monoclonal Antibodies for the Treatment of Solid Tumors" Encyclopedia, https://encyclopedia.pub/entry/41252 (accessed June 25, 2026).
Blanco, E., Chocarro, L., Fernández-Rubio, L., Bocanegra, A., Arasanz, H., Echaide, M., Garnica, M., Piñeiro-Hermida, S., Kochan, G., & Escors, D. (2023, February 15). Monoclonal Antibodies for the Treatment of Solid Tumors. In Encyclopedia. https://encyclopedia.pub/entry/41252
Blanco, Ester, et al. "Monoclonal Antibodies for the Treatment of Solid Tumors." Encyclopedia. Web. 15 February, 2023.
Copy Citation
Immunotherapies based on immune checkpoint blockade have shown remarkable clinical outcomes and durable responses in patients with many tumor types. These therapies lack efficacy in most cancer patients, even causing severe adverse events in a small subset of patients, such as inflammatory disorders and hyper-progressive disease. To diminish the risk of developing serious toxicities, intratumor delivery of monoclonal antibodies could be a solution. Encouraging results have been shown in both preclinical and clinical studies.
monoclonal antibodies (mAbs)
intratumoral therapy
viral therapy
1. Therapeutic Antibodies: Beyond Conventional Monoclonal Antibodies (mAbs)
In 1988, Greg Winter and his team pioneered the technique to humanize monoclonal antibodies, and since then the field of personalized therapy using mAbs has been successfully developed for the treatment of various cancers [1]. To date, approximately more than 100 mAbs have been approved by the US Food and Drug Administration (FDA) for the treatment of cancer [2].
Monoclonal antibodies are engineered to specifically bind target antigens with high affinity. These antibodies then have the potential to induce complement-dependent cytotoxicity and promote innate and adaptive immune responses depending on their target specificities [3]. MAbs are large immunoglobulin molecules, usually in the range of about 150 kDa. The standard mAb molecule is composed of four polypeptide chains, forming a Y-shaped macromolecule complex. More specifically, mAbs are made of two identical heavy chains and two identical light chains connected by interchain disulphide bonds and non-covalent interactions. The light chain and two N-terminal domains of the heavy chain comprise the antigen-binding fragment (Fab). The variable domains of the heavy (VH) and light (VL) chains form the antigen recognition domain by adopting a structure with three hypervariable loops in each domain that comprise the complementary-determining regions (CDRs 1, 2, and 3), flanked by four conserved framework regions (FRs 1, 2, 3, and 4) (Figure 1A). The immune-mediated functions, such as antibody-dependent cellular cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC), are conferred by the C-terminal halves of the heavy chains, which comprise the crystallizable fragment (Fc). The conserved domains of the heavy chains differ between distinct antibody classes (isotypes). From these, the immunoglobulin G (IgG) isotypes are the main ones in human plasma. Hence, IgG-based monoclonal antibodies (mAbs) have become a dominant class of biotherapeutics in recent decades [4].
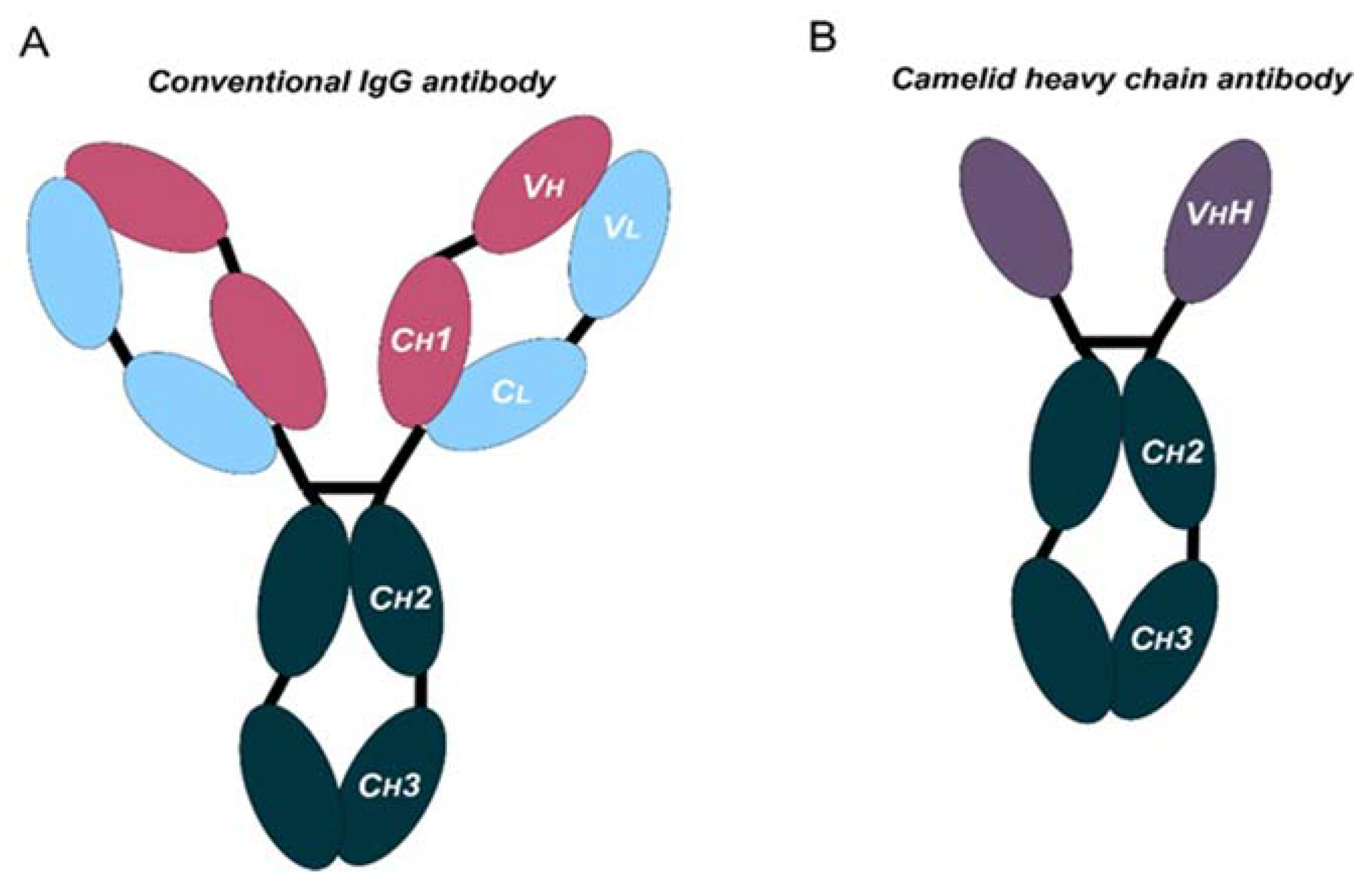
Figure 1. Schematic representation of the structure of conventional and camelid heavy-chain antibodies. (A) Structure of a conventional IgG antibody, composed by two heavy and two light chains. Conserved and variable heavy-chain domains (Ch, Vh) and light domains (Cl, Vl) are indicated. (B) Structure of a camelid heavy-chain antibody, composed by two heavy chains.
In the last decade, mAbs that block immune checkpoint molecules have shown remarkable clinical outcomes and durable responses in treated cancer patients. These mAbs have been engineered to reactivate antitumor immunity by blockade of T cell inhibitory molecules (immune checkpoints, IC), such as CTLA-4, PD-1, PD-L1, and LAG-3, which are the most studied IC. These molecules are expressed in different immune or non-immune cells. PD-L1 can be expressed by cancer cells and myeloid cells [5]. In addition, antigen-presenting cells (APC) express CTLA-4, and T cells and NK cells express PD-1, LAG-3, and other molecules, such as TIM-3 and TIGIT [6].
Antibodies targeting these latter targets are widely used for the management of cancer [7][8][9][10][11][12][13]. These therapies rely on systemic administration of these mAbs every three to four weeks. Moreover, these mAbs frequently present poor tissue penetration, intrinsic immunogenicity, and high production costs. All these issues suggest that there is still room for improvement. In fact, the large and complex structure of these mAbs causes several disadvantages: (1) high production cost due to their complexity and posttranslational modifications, (2) limited physicochemical stability, (3) systemic administration, which may cause off-target immune-related toxicities, (4) low penetration in solid tumors, and (5) poor penetration in the brain [14][15]. In certain cases, the production and use of full-length antibodies may be problematic, as mentioned above. Recent advances in antibody engineering have facilitated the production of a collection of antibody variants for the use in cancer, including other antibody formats (e.g., antibody fragments, bispecific antibodies (BsAbs), and non-IgG scaffold proteins) and antibody derivatives (e.g., antibody–drug conjugates (ADCs) and immunocytokines) [16]. Right now, the field of antibodies is revolving around the engineering of the antigen-binding region from heavy single-chain antibodies (HCAbs), such as those from camelids (dromedaries, camels, llamas, alpacas, guanacos, and vicuñas), coined with the term “nanobodies” [17]. Compared to conventional mAbs, HCAbs are homodimers made of two identical heavy-chain molecules, lacking both the light chain and the constant domain 1 (CH1) of the heavy chain. These antibodies possess a significantly smaller molecular mass, of approximately 95 kDa (Figure 1B). The variable antigen-binding domain of HCAbs (called VHH) retains full antigen-binding potential despite lacking the light chain. Indeed, the VHH domain is the smallest naturally occurring antigen-binding fragment [18].
The discovery of HCAbs has spurred the development of nanobody production platforms due to their inherent properties, which make them very attractive tools for cancer treatment: (1) nanoscale dimensions enable deeper tumor penetration, (2) certain nanobodies can cross the blood–brain barrier (BBB) [19], and (3) high affinity and specificity for their targets with low off-target accumulation. Structurally, nanobodies are similar to the VH domain of conventional antibodies, with four FRs and three CDRs. Besides, nanobodies exhibit high homology with the VH domain of human family III immunoglobulins. In addition, nanobodies present higher hydrophilicity, with increased solubility and enhanced physicochemical stability compared to the variable fragments from conventional antibodies. Moreover, the CDR3 motif tends to be longer, being the main region implicated in antigen binding. The antigen-binding region is made by CDR3 and CDR2, together with some FR residues. This extended CDR3 loop allows binding to small cavities or concave epitopes (mainly conformational epitopes), such as catalytic sites of enzymes. In contrast, conventional antibodies are better at recognizing small chemical groups (haptens), peptides, or flat epitopes on proteins [20][21]. Currently, several nanobody-based therapeutics are under clinical trials for the treatment of a variety of diseases, including cancer, autoimmune diseases, and viral infections [22].
2. Routes of mAbs Administration
The success of immune checkpoint blockade immunotherapies has demonstrated significant advances in the treatment of many cancers. However, current immunotherapies fail in most cancer patients. There are many reasons for the failure of immunotherapies, which include poor immunogenicity characterized by reduced tumor infiltration with immune cells, and systemic immune dysfunctionality [9][23][24][25]. The lack of penetration of mAbs within the tumor environment adds up to the poor immunogenicity of many cancer types. These problems contribute to the failure of ICB therapies in many cancer patients.
The systemic parenteral use of therapeutic mAbs has unequivocal advantages, allowing simplicity of administration and predictable serum pharmacokinetics. However, this mode of delivery presents limitations and disadvantages, which include poor penetration into solid tumors and systemic toxicities caused by off-target effects, with systemic inflammation and autoimmune or autoimmune-like reactions [26][27]. Indeed, serious immune-related adverse events (irAEs) are associated with recurrent systemic administration of ICB antibodies [28][29][30]. These constraints could be overcome, or at least reduced, by enhancing mAbs availability within the tumor microenvironment (TME). One way to achieve this would be through intratumor delivery of mAbs and locoregional delivery.
Several routes have been tested for mAbs delivery, for example subcutaneous (SC), intramuscular (IM), oral, and intratumor (I.T) administration. SC injection would be the most convenient for patients. Thus, various mAbs have emerged, designed for subcutaneous administration [31][32]. This administration route would be suited for self-administration by the patient, although so far, the accuracy and efficacy of mAbs delivered in such a way is hard to predict. Indeed, this is a major issue for human therapy. In addition, the mAb formulation itself for SC delivery is still a major challenge for drug development [33]. The same arguments can be applied for muscular delivery. Nevertheless, the clinical application of these type of drugs for these administration routes is hampered by their complex structure. Oral administration of mAbs has been discarded because of inefficient transport through tissue barriers such as the intestinal mucosa, while they are quickly degraded by proteases in the gastrointestinal tract. In contrast to other strategies, intratumor delivery can increase the therapeutic index of mAbs by restricting them to within the tumor environment, with a reduced risk for off-target toxicities. In addition, repeated intratumor injections can be administered, leading to much higher local bioactive drug concentrations [34][35].
Despite the challenges associated with intratumor delivery, it has substantial potential to improve immunotherapies.
3. Intratumor mAbs Delivery in Solid Tumors
Intratumor administration using image-guided injection is achievable for most organs [34]. Following administration, the therapeutic agents first diffuse throughout the injected area, thereby achieving a very high local concentration. Overtime, drugs will dissipate into systemic circulation. In fact, this gradual absorption into the blood can have pharmacokinetic advantages that will permit higher local doses with better tolerability, as shown for other protein-based drugs [36] (NCT02304393). Importantly, intratumor delivery allows the immediate access to tumor-draining lymph nodes and other lymphoid structures within the tumor tissue, amplifying the immune response [37][38][39]. Importantly, intratumor administration of mAbs and other drugs shows an abscopal effect in distal metastases, indicating that this administration route can have systemic activities [40][41]. Injections in multiple tumor lesions within the same patient can also enhance polyclonal responses, despite the high variability of cancer cells in metastases [42]. In addition, several studies have evaluated intratumoral mAbs and short peptides against receptors expressed on the blood–brain barrier (BBB) delivery to achieve brain targeting [43][44].
Several drug combinations have been evaluated with success in preclinical studies, for example combinations of antibodies targeting CTLA4, OX40, PD-1, and CD137 [45]. Some formulations also allow a slow intratumor release of mAbs, which leads to a prolonged and improved therapeutic index [46].
There are two main vector strategies for optimized intratumor mAbs administration: non-viral- and viral-based vectors. Both strategies present intrinsic advantages and disadvantages (Figure 2). The efficiency of modifying host cells with mAb-encoding nucleic acids is better with viral vectors, but a major drawback of viral vectors is their immunogenicity and potential cytotoxicity [47][48]. Replicative oncolytic viruses are also emerging as promising anti-cancer treatments, but in this case, these can be regarded as an in-situ treatment that releases antigens and damage-associated molecules, rather than virus-based vectors for drug delivery. On the other hand, non-viral vectors possess better safety profiles but less efficient capacities to modify target cells in vivo [49][50].
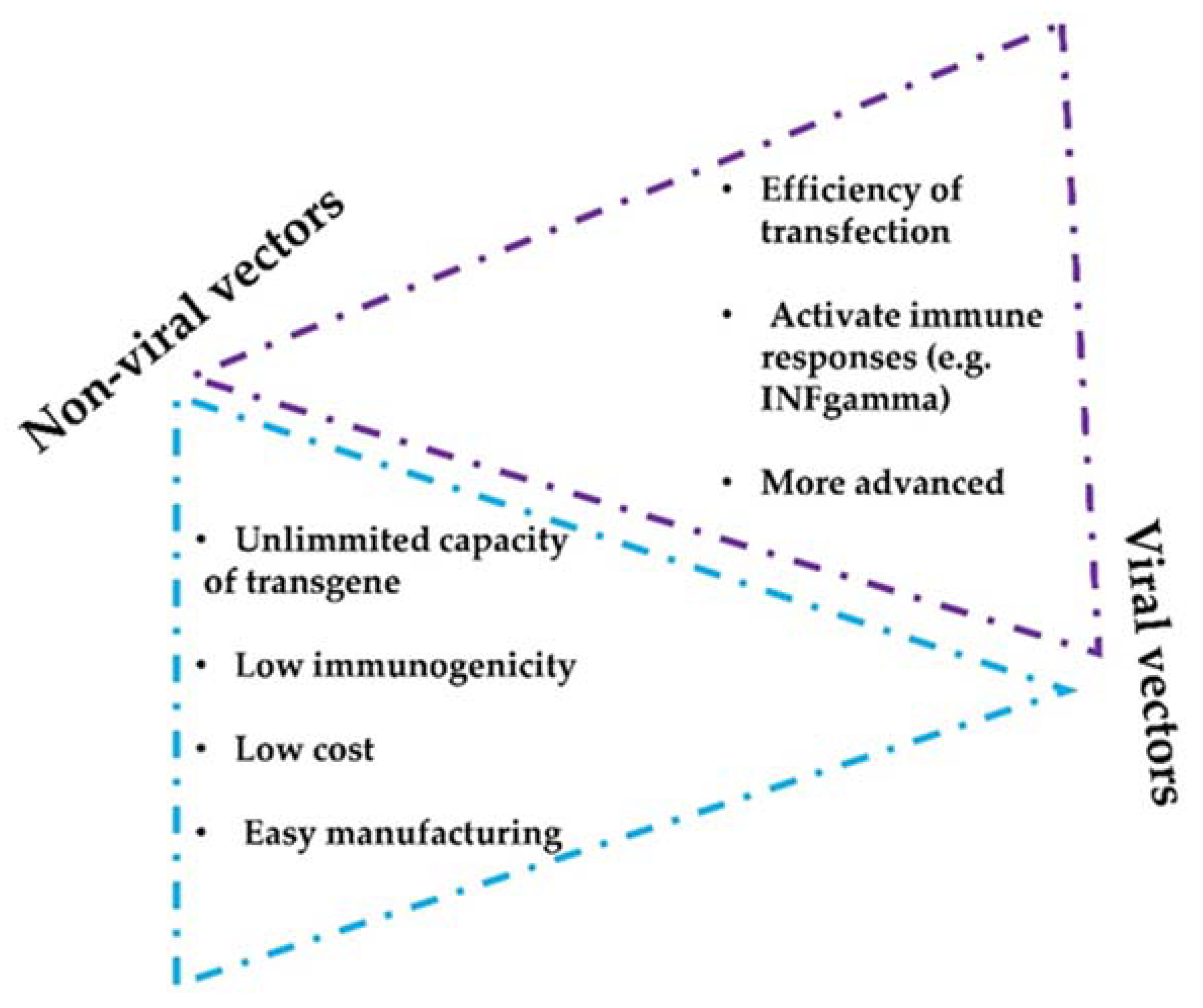
Figure 2. Considerations of non-viral and viral vectors for mAbs intratumor delivery systems. The figure shows the main advantages and limitations that distinguish viral and non-viral strategies.
Several preclinical and clinical trials are testing strategies to deliver mAbs locally within the tumor. This research focuses on the current preclinical and clinical achievements in mAbs intratumoral delivery using non-viral and viral vectors for the treatment of solid tumors.
4. Preclinical Non-Viral Vectors for mAbs Intratumor Delivery
There are a variety of delivery vehicles and scaffolds that have been engineered over time to transport biomolecules, including mAbs [51][52]. All are designed based on enhancing biodistribution within the tumors, and some have been adapted to operate within the physicochemical properties of the TME, such as a low pH or high concentrations of ATP, while sustaining their cargo release.
4.1. Nanoparticles and Lipid Vesicles
The most widely used carriers include polymer nanoparticles (NPs), inorganic NPs, and lipid-based NPs for drug delivery. The main goal for utilizing NPs is to improve the bioavailability of immunotherapeutic agents while reducing toxicity. Typically, lipid nanoparticle formulations are composed of pH-responsive lipids or cationic lipids bearing tertiary or quaternary amines to encapsulate the polyanionic RNA molecules. In addition to this main composition, nanoparticles incorporate other neutral helper lipids to constitute a hydrophilic layer over the nanoparticles to stabilize the lipid bilayer and enhance RNA delivery [53].
MnCaCO3/ICG nanoparticles have been produced loaded with PD-L1-targeted siRNA that can be intravenously injected. PD-L1 is one of the main T-cell inhibitory molecules expressed by cancer cells, through biding to PD-1 expressed on the T cell surface [54][55]. In addition, PD-L1 expression also confers cancer cells with resistance to apoptosis. Hence, PD-L1 silencing combined with photodynamic therapy (PDT) showed powerful antitumor effects [56]. These nanoparticles for the treatment of cancer have been used to treat recurrence after surgical resection. A fibrin gel was used to encapsulate calcium carbonate nanoparticles pre-loaded with anti-CD47 antibody and applied locally in the tumor. This treatment achieved polarization of tumor-associated macrophages towards M1-like phenotypes, leading to tumor control both locally and distally after surgery [57]. Another interesting strategy is based on the use of PD-1-positive tumor-derived vesicles to disrupt PD-1/PD-L1 interactions [58]. Recently, it has been shown that PD-L1 present on tumor cell-derived extracellular vesicles (sEVs) play a key role in immunosuppression and resistance to immunotherapies. Therefore, counteracting these vesicles could also improve conventional treatments [59].
Moreover, polyethylenimine (PEI) is a cationic polymer that has been extensively used for intratumoral delivery. PEI can be modified with cholesterol or other lipoic acids to improve gene delivery. In fact, a potent nanoplexed formulation with Poly I:C complexed with PEI was recently developed. The powerful antitumoral activity in murine models led to clinical evaluation [60] (NCT02828098).
4.2. Microneedle Delivery Platforms
Microneedles (MNs) have become a leading delivery strategy for transdermal drug administration and have been reconverted for immunotherapies. MNs are micron-sized and minimally invasive. These microneedles facilitate transdermal local delivery of different cargoes, from proteins to small molecules. This procedure achieves controlled and sustained cargo release [61]. Microneedle (MN) patches can also be formulated to modulate the TME, for example biodegradable and pH-sensitive MNs, or the MN-based GOx/CAT enzymatic system. Wang and colleagues developed a new procedure to perform localized delivery of anti-PD-1 for melanoma treatment, in which MN was integrated with pH-sensitive dextran nanoparticles loaded with glucose oxidase (GOx). This mechanism of delivery is PH-dependent. A decrease in pH promotes self-degradation of the nanoparticles within the MN, allowing a continuous release of mAb within the tumor environment. This intratumoral strategy showed efficacious tumor growth inhibition in vivo in a mouse melanoma model [62].
In addition, MN tools allow intratumoral co-delivery of two or more ICB agents to achieve synergistic therapeutic effects. MNs based on the GOx/CAT enzymatic system facilitated sustained release of ICB therapeutics, for example to block PD-1 and deliver an indoleamine 2,3-dioxygenase (IDO) inhibitor in a B16F10 mouse melanoma tumor model. A synergistic anti-tumor activity of IDO inhibition and PD-1 blockade was observed with prolonged survival [63]. Modifications of MN strategies can be performed with other approaches, for example implementation of MNs implemented with cold atmospheric plasma (CAP) to facilitate transdermal penetration of CAP to tumor tissues to induce immunogenic death. In this way, enhanced release of tumor-associated antigens was achieved to elicit dendritic cell maturation (DC) and T cell responses [64].
4.3. Hydrogels as Delivery Vehicles
Hydrogels are biomaterials formed by a cross-linked porous network of polymers. Some of these are termed ‘smart biomaterials’ when they have the property of changing their structural properties to respond to environmental stimuli (e.g., light, temperature, pressure, electric and/or magnetic fields, pH, solvent composition, and recognition of ions and specific molecules) [65][66][67]. Hydrogels have been proven to be non-toxic and biodegradable, becoming a potential vehicle for encapsulating therapeutic molecules. It has recently been shown in preclinical murine melanoma and breast cancer models that a PEG-b-poly(L-alanine) hydrogel permitted encapsulation and release of tumor lysate cells with granulocyte–macrophage colony stimulating factor (GMCSF), anti-PD-1, and anti-CTLA-4 simultaneously in the tumor. This co-delivery of a tumor vaccine and dual immune checkpoint inhibitors showed a significant increase of efficacy [68]. Combinatorial local immunotherapy with celecoxib and anti-PD-1 from a hydrogel system synergistically enhanced activated T cells, and reduced regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) within the tumor microenvironment [69].
4.4. HSC−Platelet−Anti-PD-1 Assembly
Platelet engineering has surfaced as an interesting novel approach due to their unique targeting ability toward inflammation sites. Natural platelets have been shown to be conjugated with anti-PD-1 antibody for targeted delivery following tumor resection to inhibit tumor recurrence. A cell–cell combinatorial delivery platform was constructed based on conjugates of platelets and hematopoietic stem cells (HSCs) for leukemia treatment. With the homing ability of HSCs to the bone marrow, the HSC–platelet–anti-PD-1 assembly could effectively deliver the anti-PD-1 antibody in an acute myeloid leukemia mouse model [70].
4.5. Intratumor Plasmid DNA (pDNA) Electroporation
An emerging strategy to be applied in human therapy is electroporation of plasmid DNA. pDNA-based delivery is cost-efficient, allows for combination therapies, and presents low immune-related toxicity risks by intratumor gene electrotransfer. This strategy has been used for the combined delivery of plasmids encoding IL-12 and an anti-PD-1 antibody that induced good anti-tumor responses [71]. Other studies combined anti-CTLA-4 and anti-PD-1 antibodies to evaluate their pharmacokinetics and pharmacodynamics when delivered via intramuscular or intratumor electroporation in mice [72].
4.6. Antigen Peptides Conjugated on mAbs
New strategies based on engineered mAbs are appearing. Indeed, antigen/α-PD-L1 conjugate therapy showed a strong local antitumor immune response [73]. In line with similar works, an anti-PD-L1 peptide-conjugated prodrug nanoparticle (PD-NP) has been developed to avoid severe toxicity and improve antitumor activity of T cells in cancer immunotherapy [74].
References
- Buss, N.A.; Henderson, S.J.; McFarlane, M.; Shenton, J.M.; de Haan, L. Monoclonal antibody therapeutics: History and future. Curr. Opin. Pharmacol. 2012, 12, 615–622.
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2021. mAbs 2021, 13, 1860476.
- Jin, S.; Sun, Y.; Liang, X.; Gu, X.; Ning, J.; Xu, Y.; Chen, S.; Pan, L. Emerging new therapeutic antibody derivatives for cancer treatment. Signal Transduct. Target. Ther. 2022, 7, 39.
- Goulet, D.R.; Atkins, W.M. Considerations for the Design of Antibody-Based Therapeutics. J. Pharm. Sci. 2020, 109, 74–103.
- Bocanegra, A.; Blanco, E.; Fernandez-Hinojal, G.; Arasanz, H.; Chocarro, L.; Zuazo, M.; Morente, P.; Vera, R.; Escors, D.; Kochan, G. PD-L1 in Systemic Immunity: Unraveling Its Contribution to PD-1/PD-L1 Blockade Immunotherapy. Int. J. Mol. Sci. 2020, 21, 5918.
- Deshpande, R.P.; Sharma, S.; Watabe, K. The Confounders of Cancer Immunotherapy: Roles of Lifestyle, Metabolic Disorders and Sociological Factors. Cancers 2020, 12, 2983.
- Edwards, C.J.; Sette, A.; Cox, C.; Di Fiore, B.; Wyre, C.; Sydoruk, D.; Yadin, D.; Hayes, P.; Stelter, S.; Bartlett, P.D.; et al. The multi-specific VH-based Humabody CB213 co-targets PD1 and LAG3 on T cells to promote anti-tumour activity. Br. J. Cancer 2022, 126, 1168–1177.
- Chocarro, L.; Bocanegra, A.; Blanco, E.; Fernandez-Rubio, L.; Arasanz, H.; Echaide, M.; Garnica, M.; Ramos, P.; Pineiro-Hermida, S.; Vera, R.; et al. Cutting-Edge: Preclinical and Clinical Development of the First Approved Lag-3 Inhibitor. Cells 2022, 11, 2351.
- Zuazo, M.; Arasanz, H.; Fernandez-Hinojal, G.; Garcia-Granda, M.J.; Gato, M.; Bocanegra, A.; Martinez, M.; Hernandez, B.; Teijeira, L.; Morilla, I.; et al. Functional systemic CD4 immunity is required for clinical responses to PD-L1/PD-1 blockade therapy. EMBO Mol. Med. 2019, 11, e10293.
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P.; et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552.
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454.
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465.
- O’Day, S.J.; Maio, M.; Chiarion-Sileni, V.; Gajewski, T.F.; Pehamberger, H.; Bondarenko, I.N.; Queirolo, P.; Lundgren, L.; Mikhailov, S.; Roman, L.; et al. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: A multicenter single-arm phase II study. Ann. Oncol. 2010, 21, 1712–1717.
- Castelli, M.S.; McGonigle, P.; Hornby, P.J. The pharmacology and therapeutic applications of monoclonal antibodies. Pharmacol. Res. Perspect. 2019, 7, e00535.
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233.
- Weiner, G.J. Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 2015, 15, 361–370.
- Verhaar, E.R.; Woodham, A.W.; Ploegh, H.L. Nanobodies in cancer. Semin. Immunol. 2021, 52, 101425.
- Yang, E.Y.; Shah, K. Nanobodies: Next Generation of Cancer Diagnostics and Therapeutics. Front. Oncol. 2020, 10, 1182.
- Rissiek, B.; Koch-Nolte, F.; Magnus, T. Nanobodies as modulators of inflammation: Potential applications for acute brain injury. Front. Cell. Neurosci. 2014, 8, 344.
- Bannas, P.; Hambach, J.; Koch-Nolte, F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Front. Immunol. 2017, 8, 1603.
- Panikar, S.S.; Banu, N.; Haramati, J.; Del Toro-Arreola, S.; Riera Leal, A.; Salas, P. Nanobodies as efficient drug-carriers: Progress and trends in chemotherapy. J. Control. Release 2021, 334, 389–412.
- Jovčevska, I.; Muyldermans, S. The Therapeutic Potential of Nanobodies. BioDrugs 2020, 34, 11–26.
- Tenholder, M.F. Pulmonary infections in the immunocompromised host. Perspective on procedures. Chest 1988, 94, 676–678.
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168.
- Arasanz, H.; Bocanegra, A.I.; Morilla, I.; Fernandez-Irigoyen, J.; Martinez-Aguillo, M.; Teijeira, L.; Garnica, M.; Blanco, E.; Chocarro, L.; Ausin, K.; et al. Circulating Low Density Neutrophils Are Associated with Resistance to First Line Anti-PD1/PDL1 Immunotherapy in Non-Small Cell Lung Cancer. Cancers 2022, 14, 3846.
- Khan, Z.; Hammer, C.; Guardino, E.; Chandler, G.S.; Albert, M.L. Mechanisms of immune-related adverse events associated with immune checkpoint blockade: Using germline genetics to develop a personalized approach. Genome Med. 2019, 11, 39.
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. New Engl. J. Med. 2018, 378, 158–168.
- Darnell, E.P.; Mooradian, M.J.; Baruch, E.N.; Yilmaz, M.; Reynolds, K.L. Immune-Related Adverse Events (irAEs): Diagnosis, Management, and Clinical Pearls. Curr. Oncol. Rep. 2020, 22, 39.
- Arasanz, H.; Zuazo, M.; Bocanegra, A.; Chocarro, L.; Blanco, E.; Martinez, M.; Morilla, I.; Fernandez, G.; Teijeira, L.; Morente, P.; et al. Hyperprogressive Disease: Main Features and Key Controversies. Int. J. Mol. Sci. 2021, 22, 3736.
- Arasanz, H.; Zuazo, M.; Bocanegra, A.; Gato, M.; Martinez-Aguillo, M.; Morilla, I.; Fernandez, G.; Hernandez, B.; Lopez, P.; Alberdi, N.; et al. Early detection of hyperprogressive disease in non-small cell lung cancer by monitoring of systemic T cell dynamics. Cancers 2020, 12, 344.
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorganic Med. Chem. 2018, 26, 2700–2707.
- Bittner, B.; Richter, W.; Schmidt, J. Subcutaneous Administration of Biotherapeutics: An Overview of Current Challenges and Opportunities. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2018, 32, 425–440.
- Viola, M.; Sequeira, J.; Seica, R.; Veiga, F.; Serra, J.; Santos, A.C.; Ribeiro, A.J. Subcutaneous delivery of monoclonal antibodies: How do we get there? J. Control. Release 2018, 286, 301–314.
- Champiat, S.; Tselikas, L.; Farhane, S.; Raoult, T.; Texier, M.; Lanoy, E.; Massard, C.; Robert, C.; Ammari, S.; De Baere, T.; et al. Intratumoral Immunotherapy: From Trial Design to Clinical Practice. Clin. Cancer Res. 2021, 27, 665–679.
- Goldmacher, G.V.; Khilnani, A.D.; Andtbacka, R.H.I.; Luke, J.J.; Hodi, F.S.; Marabelle, A.; Harrington, K.; Perrone, A.; Tse, A.; Madoff, D.C.; et al. Response Criteria for Intratumoral Immunotherapy in Solid Tumors: ItRECIST. J. Clin. Oncol. 2020, 38, 2667–2676.
- Margolin, K.; Morishima, C.; Velcheti, V.; Miller, J.S.; Lee, S.M.; Silk, A.W.; Holtan, S.G.; Lacroix, A.M.; Fling, S.P.; Kaiser, J.C.; et al. Phase I Trial of ALT-803, A Novel Recombinant IL15 Complex, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2018, 24, 5552–5561.
- Petitprez, F.; de Reynies, A.; Keung, E.Z.; Chen, T.W.; Sun, C.M.; Calderaro, J.; Jeng, Y.M.; Hsiao, L.P.; Lacroix, L.; Bougouin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560.
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555.
- Sweeney, K.J.; Tetzlaff, M.T.; Vega, F.; Gillenwater, A.; Zuo, Z.; Gross, N.; Nagarajan, P.; Wargo, J.; Nelson, K.; Prieto, V.G.; et al. Tertiary lymphoid structures with overlapping histopathologic features of cutaneous marginal zone lymphoma during neoadjuvant cemiplimab therapy are associated with antitumor response. J. Cutan. Pathol. 2021, 48, 674–679.
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat Rev Cancer 2018, 18, 313–322.
- Brody, J.D.; Ai, W.Z.; Czerwinski, D.K.; Torchia, J.A.; Levy, M.; Advani, R.H.; Kim, Y.H.; Hoppe, R.T.; Knox, S.J.; Shin, L.K.; et al. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: A phase I/II study. J. Clin. Oncol. 2010, 28, 4324–4332.
- Hong, W.X.; Haebe, S.; Lee, A.S.; Westphalen, C.B.; Norton, J.A.; Jiang, W.; Levy, R. Intratumoral Immunotherapy for Early-stage Solid Tumors. Clin. Cancer Res. 2020, 26, 3091–3099.
- Lockman, P.R.; Mumper, R.J.; Khan, M.A.; Allen, D.D. Nanoparticle technology for drug delivery across the blood-brain barrier. Drug Dev. Ind. Pharm. 2002, 28, 1–13.
- Galstyan, A.; Markman, J.L.; Shatalova, E.S.; Chiechi, A.; Korman, A.J.; Patil, R.; Klymyshyn, D.; Tourtellotte, W.G.; Israel, L.L.; Braubach, O.; et al. Blood-brain barrier permeable nano immunoconjugates induce local immune responses for glioma therapy. Nat. Commun. 2019, 10, 3850.
- Marabelle, A.; Andtbacka, R.; Harrington, K.; Melero, I.; Leidner, R.; de Baere, T.; Robert, C.; Ascierto, P.A.; Baurain, J.F.; Imperiale, M.; et al. Starting the fight in the tumor: Expert recommendations for the development of human intratumoral immunotherapy (HIT-IT). Ann. Oncol. 2018, 29, 2163–2174.
- Tselikas, L.; de Baere, T.; Isoardo, T.; Susini, S.; Ser-Le Roux, K.; Polrot, M.; Adam, J.; Rouanne, M.; Zitvogel, L.; Moine, L.; et al. Pickering emulsions with ethiodized oil and nanoparticles for slow release of intratumoral anti-CTLA4 immune checkpoint antibodies. J. Immunother. Cancer 2020, 8, e000579.
- Escors, D.; Breckpot, K. Lentiviral Vectors in Gene Therapy: Their Current Status and Future Potential. Arch. Immunol. Ther. Exp. 2010, 58, 107–119.
- Breckpot, K.; Escors, D.; Arce, F.; Lopes, L.; Karwacz, K.; Van Lint, S.; Keyaerts, M.; Collins, M. HIV-1 lentiviral vector immunogenicity is mediated by Toll-like receptor 3 (TLR3) and TLR7. J. Virol. 2010, 84, 5627–5636.
- Sayed, N.; Allawadhi, P.; Khurana, A.; Singh, V.; Navik, U.; Pasumarthi, S.K.; Khurana, I.; Banothu, A.K.; Weiskirchen, R.; Bharani, K.K. Gene therapy: Comprehensive overview and therapeutic applications. Life Sci. 2022, 294, 120375.
- Hemminki, O.; Dos Santos, J.M.; Hemminki, A. Oncolytic viruses for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 84.
- Gil, C.J.; Li, L.; Hwang, B.; Cadena, M.; Theus, A.S.; Finamore, T.A.; Bauser-Heaton, H.; Mahmoudi, M.; Roeder, R.K.; Serpooshan, V. Tissue engineered drug delivery vehicles: Methods to monitor and regulate the release behavior. J. Control. Release 2022, 349, 143–155.
- Zhao, Y.; Bilal, M.; Qindeel, M.; Khan, M.I.; Dhama, K.; Iqbal, H.M.N. Nanotechnology-based immunotherapies to combat cancer metastasis. Mol. Biol. Rep. 2021, 48, 6563–6580.
- Guevara, M.L.; Persano, S.; Persano, F. Lipid-Based Vectors for Therapeutic mRNA-Based Anti-Cancer Vaccines. Curr. Pharm. Des. 2019, 25, 1443–1454.
- Karwacz, K.; Bricogne, C.; Macdonald, D.; Arce, F.; Bennett, C.L.; Collins, M.; Escors, D. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8(+) T cells. EMBO Mol. Med. 2011, 3, 581–592.
- Gato-Canas, M.; Zuazo, M.; Arasanz, H.; Ibanez-Vea, M.; Lorenzo, L.; Fernandez-Hinojal, G.; Vera, R.; Smerdou, C.; Martisova, E.; Arozarena, I.; et al. PDL1 Signals through Conserved Sequence Motifs to Overcome Interferon-Mediated Cytotoxicity. Cell Rep. 2017, 20, 1818–1829.
- Liu, Y.; Pan, Y.; Cao, W.; Xia, F.; Liu, B.; Niu, J.; Alfranca, G.; Sun, X.; Ma, L.; de la Fuente, J.M.; et al. A tumor microenvironment responsive biodegradable CaCO(3)/MnO(2)- based nanoplatform for the enhanced photodynamic therapy and improved PD-L1 immunotherapy. Theranostics 2019, 9, 6867–6884.
- Chen, Q.; Wang, C.; Zhang, X.; Chen, G.; Hu, Q.; Li, H.; Wang, J.; Wen, D.; Zhang, Y.; Lu, Y.; et al. In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment. Nat. Nanotechnol. 2019, 14, 89–97.
- Zhang, X.; Wang, C.; Wang, J.; Hu, Q.; Langworthy, B.; Ye, Y.; Sun, W.; Lin, J.; Wang, T.; Fine, J.; et al. PD-1 Blockade Cellular Vesicles for Cancer Immunotherapy. Adv. Mater. 2018, 30, e1707112.
- Yu, Z.L.; Liu, J.Y.; Chen, G. Small extracellular vesicle PD-L1 in cancer: The knowns and unknowns. NPJ Precis. Oncol. 2022, 6, 42.
- Aznar, M.A.; Planelles, L.; Perez-Olivares, M.; Molina, C.; Garasa, S.; Etxeberría, I.; Perez, G.; Rodriguez, I.; Bolaños, E.; Lopez-Casas, P.; et al. Immunotherapeutic effects of intratumoral nanoplexed poly I:C. J. Immunother. Cancer. 2019, 7, 116.
- Lee, K.; Goudie, M.J.; Tebon, P.; Sun, W.; Luo, Z.; Lee, J.; Zhang, S.; Fetah, K.; Kim, H.J.; Xue, Y.; et al. Non-transdermal microneedles for advanced drug delivery. Adv. Drug Deliv. Rev. 2020, 165–166, 41–59.
- Wang, C.; Ye, Y.; Hochu, G.M.; Sadeghifar, H.; Gu, Z. Enhanced Cancer Immunotherapy by Microneedle Patch-Assisted Delivery of Anti-PD1 Antibody. Nano Lett. 2016, 16, 2334–2340.
- Ye, Y.; Wang, J.; Hu, Q.; Hochu, G.M.; Xin, H.; Wang, C.; Gu, Z. Synergistic Transcutaneous Immunotherapy Enhances Antitumor Immune Responses through Delivery of Checkpoint Inhibitors. ACS Nano 2016, 10, 8956–8963.
- Yan, D.; Sherman, J.H.; Keidar, M. Cold atmospheric plasma, a novel promising anti-cancer treatment modality. Oncotarget 2017, 8, 15977–15995.
- Jiang, Y.; Wang, Y.; Li, Q.; Yu, C.; Chu, W. Natural Polymer-based Stimuli-responsive Hydrogels. Curr. Med. Chem. 2020, 27, 2631–2657.
- Kahn, J.S.; Hu, Y.; Willner, I. Stimuli-Responsive DNA-Based Hydrogels: From Basic Principles to Applications. Acc. Chem. Res. 2017, 50, 680–690.
- Chen, Z.; Chen, Y.; Hedenqvist, M.S.; Chen, C.; Cai, C.; Li, H.; Liu, H.; Fu, J. Multifunctional conductive hydrogels and their applications as smart wearable devices. J. Mater. Chem. B 2021, 9, 2561–2583.
- Song, H.; Yang, P.; Huang, P.; Zhang, C.; Kong, D.; Wang, W. Injectable polypeptide hydrogel-based co-delivery of vaccine and immune checkpoint inhibitors improves tumor immunotherapy. Theranostics 2019, 9, 2299–2314.
- Li, Y.; Fang, M.; Zhang, J.; Wang, J.; Song, Y.; Shi, J.; Li, W.; Wu, G.; Ren, J.; Wang, Z.; et al. Hydrogel dual delivered celecoxib and anti-PD-1 synergistically improve antitumor immunity. Oncoimmunology 2016, 5, e1074374.
- Han, X.; Li, H.; Zhou, D.; Chen, Z.; Gu, Z. Local and Targeted Delivery of Immune Checkpoint Blockade Therapeutics. Acc. Chem. Res. 2020, 53, 2521–2533.
- Jacobs, L.; Yshii, L.; Junius, S.; Geukens, N.; Liston, A.; Hollevoet, K.; Declerck, P. Intratumoral DNA-based delivery of checkpoint-inhibiting antibodies and interleukin 12 triggers T cell infiltration and anti-tumor response. Cancer Gene Ther. 2022, 29, 984–992.
- Jacobs, L.; De Smidt, E.; Geukens, N.; Declerck, P.; Hollevoet, K. DNA-Based Delivery of Checkpoint Inhibitors in Muscle and Tumor Enables Long-Term Responses with Distinct Exposure. Mol. Ther. 2020, 28, 1068–1077.
- Lee, E.J.; Jang, G.Y.; Lee, S.E.; Lee, J.W.; Han, H.D.; Park, Y.M.; Kang, T.H. A novel form of immunotherapy using antigen peptides conjugated on PD-L1 antibody. Immunol. Lett. 2021, 240, 137–148.
- Moon, Y.; Shim, M.K.; Choi, J.; Yang, S.; Kim, J.; Yun, W.S.; Cho, H.; Park, J.Y.; Kim, Y.; Seong, J.K.; et al. Anti-PD-L1 peptide-conjugated prodrug nanoparticles for targeted cancer immunotherapy combining PD-L1 blockade with immunogenic cell death. Theranostics 2022, 12, 1999–2014.
More
Information
Subjects:
Immunology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
2 times
(View History)
Update Date:
16 Feb 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No